6/6) The Crystallographic Information File - CIF
We pick up from the end of
part 4 or
part 5 of this tutorial.
The
CIF written by
SHELXL must be edited before it can accompany a
manuscript, or be deposited in a database. This is because some information is missing,
and some is incorrect. This final part of the sucrose tutorial shows how to edit the
CIF made by
SHELXL so that it passes the IUCr structure checking program
checkCIF.
The IUCr (International Union of Crystallography) has another excellent tool,
publCIF,
for editing and checking
CIFs, but in this tutorial we will not use it. If you
intend to publish in an IUCr journal, it really pays to use
publCIF as it is
used by the Acta Crystallographica journals to typeset structure-related papers.
You should have your
CIF from the last
SHELXL job
(
part 4 of this tutorial). If you don't
have it, you can download this copy:
sucrose.cif.
There is also a fully edited version of this
CIF,
sucrose-edited.cif, for you to
inspect. Without getting into the arcana of the
CIF format, the easiest way to
get started is to compare a newly generated
CIF with a fully edited version.
Warning: The
CIF format is finicky. Misplaced semi-colons are a particular
source of grief, as is the legacy 80 character per line limit.
Note: This tutorial
does not show you how to construct a full report on
your crystal structure, it merely shows you how to correct the problems with a
SHELXL-generated
CIF.
Start by opening the
CIF in your text editor. As with editing
SHELX files,
do
not use a word processor. In this tutorial you'll see successive portions of
the
CIF. If you roll over the image with your mouse it will highlight the lines
that need to be edited in red. Each of these images will be followed by a brief
description of the changes needed, followed by an image of the corresponding section of
the
CIF after it has been edited.
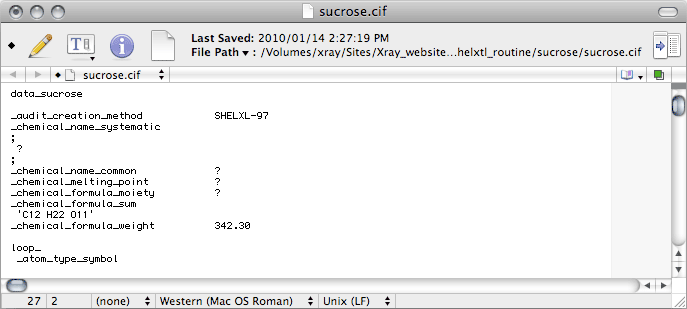
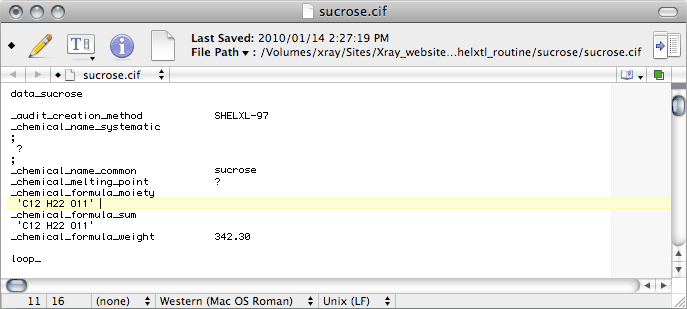
Some crystal structures have multiple fragments in the asymmetric unit, such as ions,
solvent molecules, high Z' etc., and the _chemical_formula_moiety records
the formulae of the various pieces (moieties). For sucrose it is just the same
as the _chemical_formula_sum, so just duplicate the _chemical_formula_sum
line. If you don't know what the proper moiety formula is, don't enter it yet.
Later, the output from checkCIF will tell you what it is. If you happen
to know the answer to any of the other items here, you should replace the ? with the
correct response.
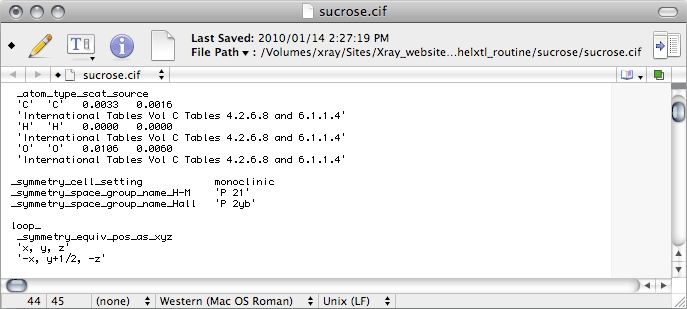
Scrolling down the CIF we come to this:
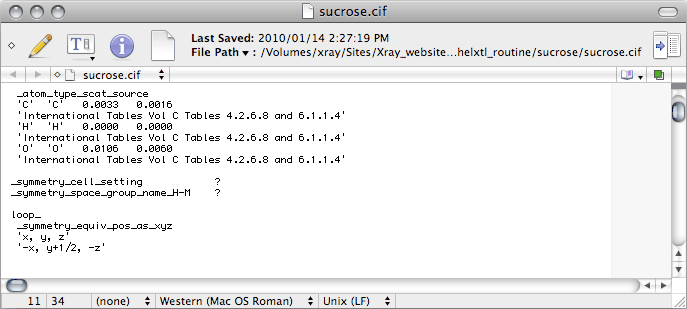
_symmetry_cell_setting is where you enter the crystal system, for sucrose this
is monoclinic.
_symmetry_space_group_name_H-M is where you enter the Hermann-Mauguin
space group symbol, here it is 'P 21', which is CIF-ese for
P21.
There are other _symmetry_space_group_ CIF data names. For some strange
reason it seems to have become common practice to enter Hall's otherwise little-used
space-group notation, which for this space group is P2yb. Hall's nomenclature is
not so well known, so if you don't know the Hall symbol for your space group, don't
enter it yet. Later, the output from checkCIF will tell you what it should be.
For sucrose it is:
_symmetry_space_group_name_Hall 'P 2yb'
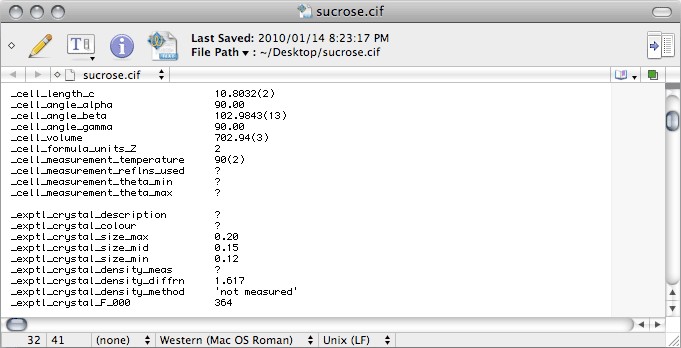
Further down in the CIF we have this:
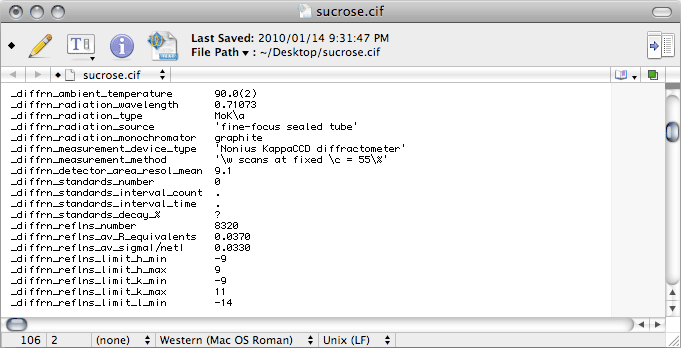
The
number of reflections,
theta_min and
theta_max used for the
cell determination are given in the
nreport file.
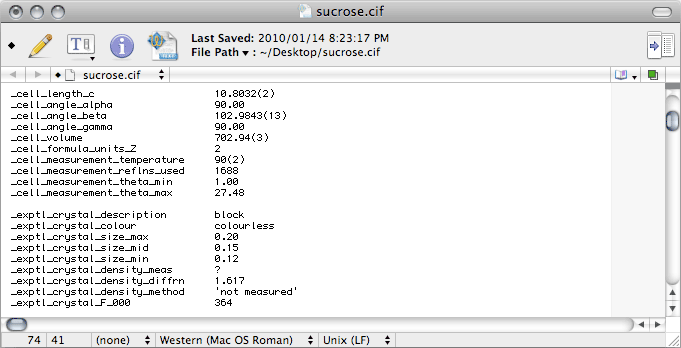
The
_exptl_crystal_description just describes the crystal shape (
e.g.
block, plate, needle
etc.), and its colour should be obvious. If you have
forgotten these details, let this be a reminder to
keep good notes.
On to the next section ...
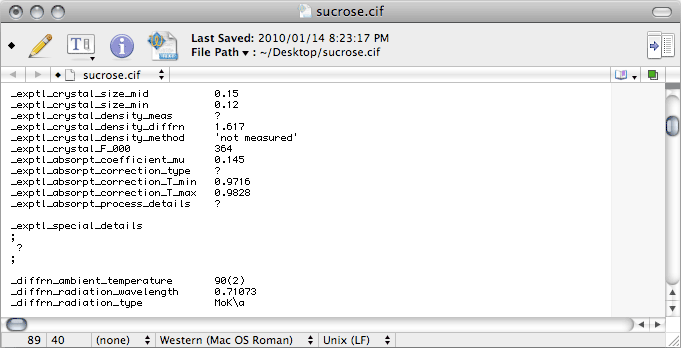
The most common type of absorption correction nowadays is the multi-scan method. This
is typically done with SADABS (by George Sheldrick), SORTAV (by Bob
Blessing) or Scalepack (by Zbyszek Otwinowski). Unfortunately, although
Scalepack is an excellent program, it does not give you the proper information
to edit this section of the CIF, so it is probably better to use SADABS
or SORTAV.
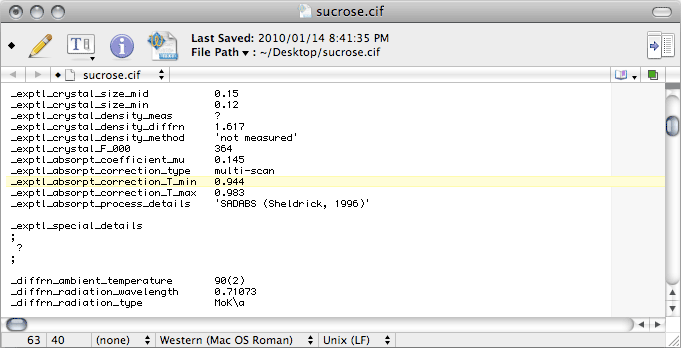
Notice that the
T_min and
T_max values are rounded to three decimal
places, and that
T_min is a little different from the value in the unedited
file. The problem is that the unedited value is not derived from the absorption
correction, it comes from a simple calculation based on crystal size (
SIZE
command in
SHELXL) and chemical composition. If
Scalepack was used for
merging, that is the best estimate you have. For
SADABS data though, the proper
value requires you to look in the
SADABS log (the
default name of this file is
sad.abs).
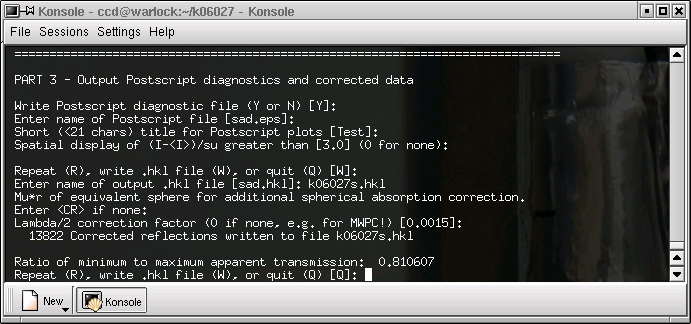
Near the end of
sad.abs you will find a line like this:
Ratio of minimum to maximum apparent transmission: 0.960326
The value for
_exptl_absorpt_correction_T_min is found by multiplying this
number (
i.e. 0.960326 for this dataset) by the
CIF value of
T_max
(0.9828 in this case), to give 0.944. If that seems a bit contrived, well that's because
it is a bit contrived. Nevertheless, it's the best estimate we have, so that's what we
use. Note that for analytical absorption corrections, the
T_min and
T_max
values should be output by the program used, and you'll need to edit both of these values in the
CIF.
Further down the
CIF we find this:
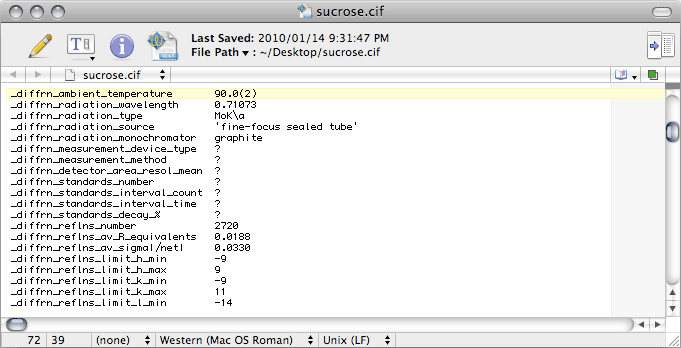
There's a bunch of stuff to change. The next image shows what the right entries should
be in this case, but other diffractometers require different information.
With area detector data, it is unusual for standard reflections to be collected, but
you should change these entries anyway. In
CIF-ese, the full stop, or period '.'
and the question mark '?' mean different things. The former means "
not applicable"
while the latter means "
not known". The difference is subtle, but you might as
well get it right.
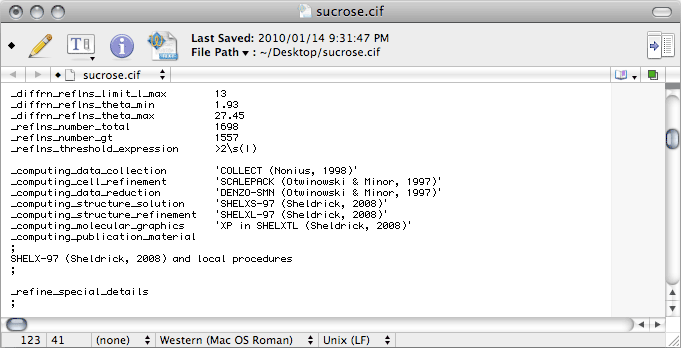
For
Scalepack, the values for
_diffrn_reflns_number and
_diffrn_reflns_av_R_equivalents are in the
nreport
file (see '
Total number of integrated reflections' and '
Overall R-merge
(linear)' in the last table), while for
SADABS look in the log file.
On to the next block ...
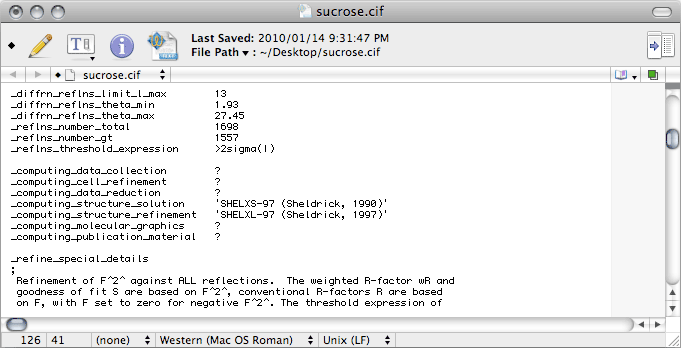
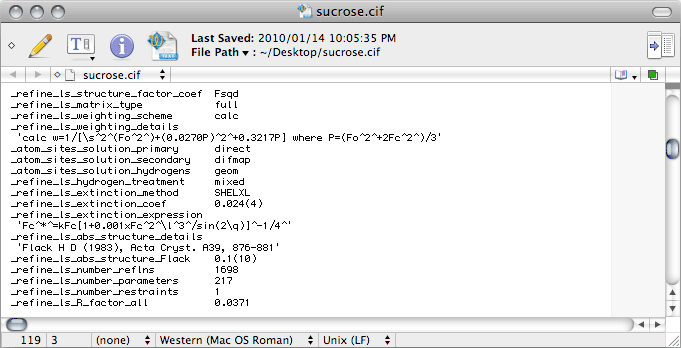
The Greek letter sigma (σ) is given as '\s' in CIF-ese. The
_computing_ entries need to show the most current references for all the programs
used. Note that if you use publCIF, this should be added on the
_computing_publication_material line. For a CIF intended for an IUCr
journal, you will also need to add a bunch of extra stuff, including
_publ_section_references, but that is beyond the scope of this tutorial. If you
are wondering what is meant by 'local procedures', it simply refers to tasks
like manual editing of the CIF.
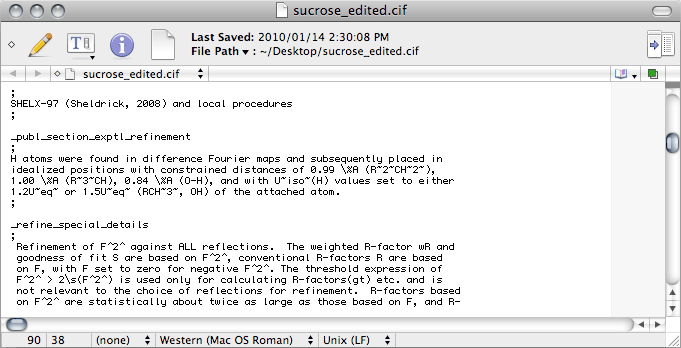
No matter what the intended purpose of the CIF, if there are hydrogen atoms in
the structure there should be a description of how the hydrogen atoms were treated.
This description goes in the _publ_ section of the CIF, but SHELXL
does not write any _publ_ lines at all. You can enter it by hand in the space
indicated by the red line in the roll-over image. While you're at it, you may as well
change the 'sigma' to '\s' in the _refine_special_details section.
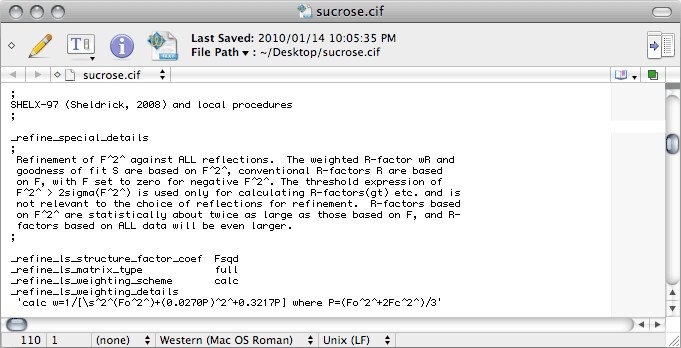
This ought to give you something like the following:
The last thing that needs editing in this first pass through the CIF is here:
By default, SHELXL writes 'mixed' on the _refine_ls_hydrogen_treatment line. This would only be appropriate if you used a combination of constrained and refined H atoms. In this sucrose example we used a riding model for all of them, so this should be changed to 'constr', which is short for constrained.
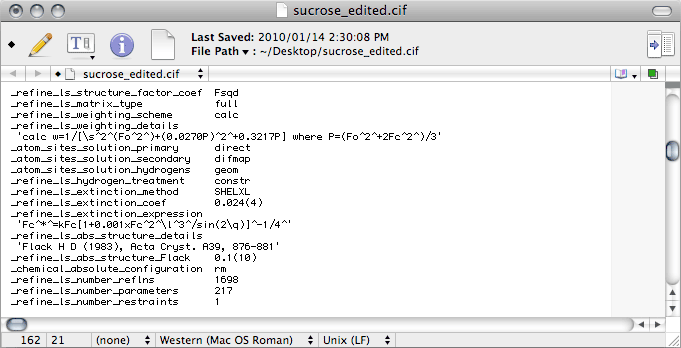
Notice that the
CIF contains the lines
_refine_ls_abs_structure_details
and
_refine_ls_abs_structure_Flack, even though you could not use Flack's
parameter to establish the absolute structure (see
part 4 of this tutorial). In a case
like this, you may be asked to remove these lines. The argument made for removing them
is that a Flack parameter with an
SU (
standard uncertainty) so large
renders the parameter 'meaningless'. In point of fact, such a value is not 'meaningless'
at all, it tells you in very definite terms that the x-ray data alone cannot establish
the absolute configuration, particularly once Friedel pairs have been merged. As such,
it confirms what we know from the physics of anomalous dispersion. This is a subtle
point that is too often lost on journal editors and referees.
You should also add a line
_chemical_absolute_configuration with the
data
value '
rm', which stands for '
reference molecule'. This states
that the handedness of your model is fixed by a reference molecule, in this case
obviously it is sucrose! If you prefer, you could put this extra line near the top of
the
CIF along with the other
_chemical_ entries, but it doesn't really
matter where it goes.
If you are lucky, your
CIF may now be sufficiently complete to survive
checkCIF.
Since check
CIF gives lots of diagnostic comments, it can be helpful for
correcting many
CIF problems. It is also useful for detecting more general
problems with your structure. So, open a browser and go to the
checkCIF
page:
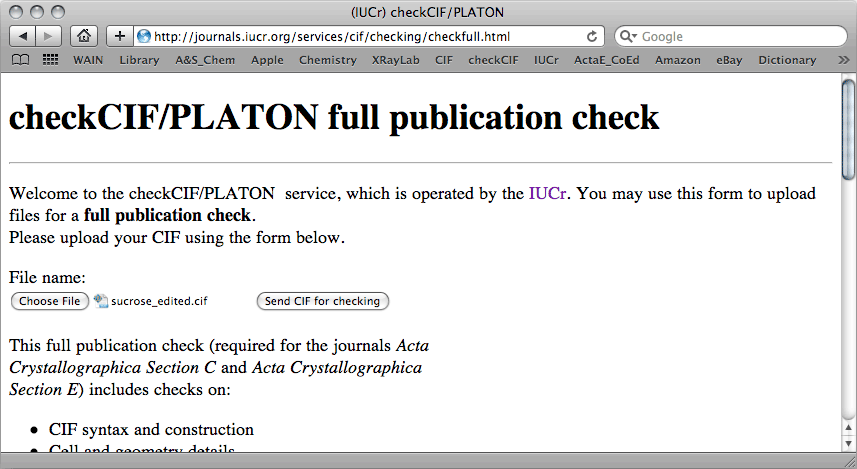
For this sucrose example, checkCIF returned the following report:
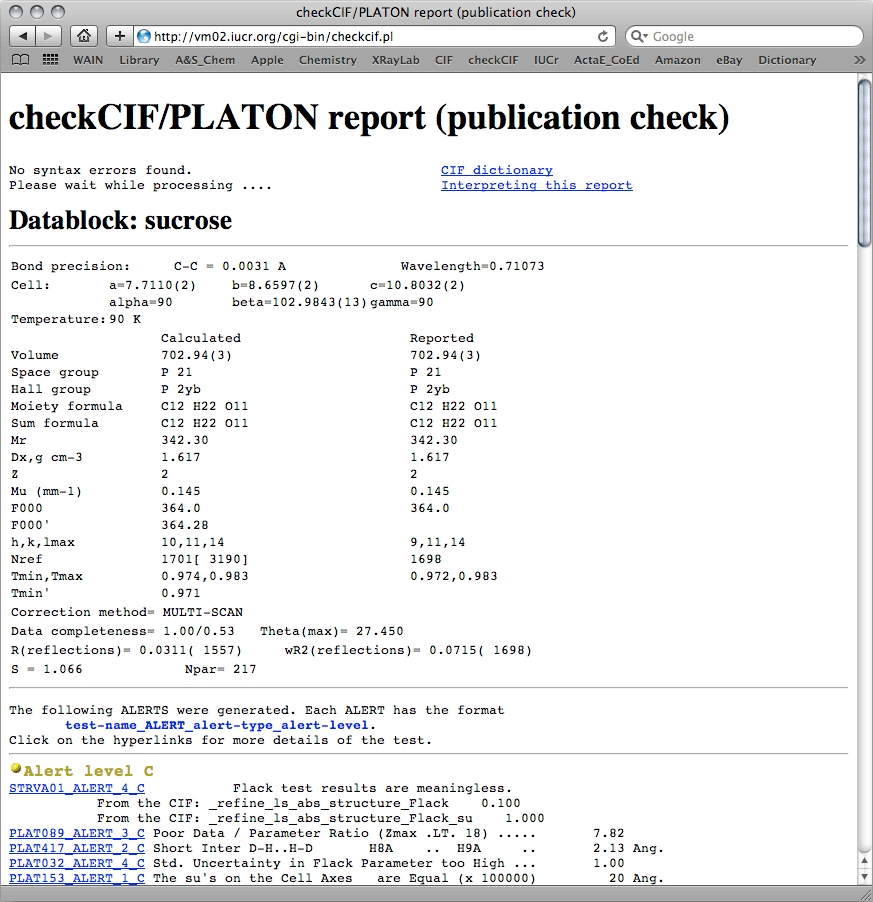
A few things here are worth mentioning. The check
CIF report shows no syntax
errors, and the table shows that
calculated and
reported data are very
similar. The differences in
Nref show that our dataset is missing three
reflections out of about 1700. That's not so bad, and it's likely just because the
program calculated
hmax = 10 rather than the
hmax
= 9 present in the dataset, and that was likely due to a rounding error. It also shows
that we merged the Friedel pairs (the number in square brackets for
NRef is the
expected unmerged number of reflections). This is also shown on the '
Data
completeness' line, and again there's nothing to worry about.
Next comes the '
Alert' section of the report. There are four levels of alert:
A, B, C, and G. Further down, the report tells you how serious these alert levels tend
to be. Generally speaking,
A-level alerts are serious and need to either be
fixed or explained in a '
Validation Reply Form'.
B-level alerts are
potentially serious, and these too should either be fixed or you should have a
well-reasoned explanation. Be aware, however, that checking programs are not infallible,
so you may sometimes see
A/B-level alerts when there is really no problem at all.
That's ok so long as you have a cogent counter argument.
C/G-level alerts usually
just tell you things that you know already, but on occasion you may need to tweak the
model (and re-refine!) to eliminate the alerts.
For this sucrose tutorial, there are no
A/B-level alerts, but there are a bunch
at
C/G-level. None of them are serious. You already know about the 'meaningless
Flack parameter' argument, and it's safe to dismiss it here. The
data/parameter
ratio is low simply because the Friedel pairs were merged, and again it's not serious
for this structure. The link in the check
CIF report, reproduced here
PLAT089_ALERT_3_C, shows the somewhat arbitrary
cut-off point for generating this alert, and it is insignificant for this structure.
A fairly close contact between H8a and H9a has been flagged, but a quick look at the
structure shows that these hydrogen atoms are fine. The uncertainty in the Flack
parameter is high, but that is to be expected; there was no discernible
anomalous signal, so the Friedel pairs were merged. Again, no big deal. Lastly, the
SUs of the cell parameters all happen to have the same value. These things
happen sometimes, and it's not a problem. Further down, the
G-level alerts give
no reason for alarm.
Conclusion: The structure determination is now complete.
{kind=link}
{kind=link}