
Structure refinement involves making improvements to the model so that it fits the
diffraction data as closely as possible. There are two main processes involved, one is
automated and the other is manual. The automated process involves using a least-squares
refinement program to make small adjustments to the atomic coordinates, displacement
(aka thermal) parameters etc. The program you will use for this is called
SHELXL. The manual steps involve either changing the model by editing the text
of the '.res' file, or by using the graphics program XP. Often the automatic
stuff is referred to as simply 'refinement', while the manual stuff gets called
'model building'.
Caution: This part of the tutorial is quite long, and it will get less hand-holdy towards the end. This is because you are expected to pick things up as you go along.
It usually takes several rounds of least-squares refinement and model building to complete a structure. Part 3 of this tutorial was really just the first model-building step. The result of that was a file, sucrose.ins, that is ready for refinement by SHELXL. Before you run SHELXL, take a look inside the sucrose.ins file:
Caution: This part of the tutorial is quite long, and it will get less hand-holdy towards the end. This is because you are expected to pick things up as you go along.
It usually takes several rounds of least-squares refinement and model building to complete a structure. Part 3 of this tutorial was really just the first model-building step. The result of that was a file, sucrose.ins, that is ready for refinement by SHELXL. Before you run SHELXL, take a look inside the sucrose.ins file:
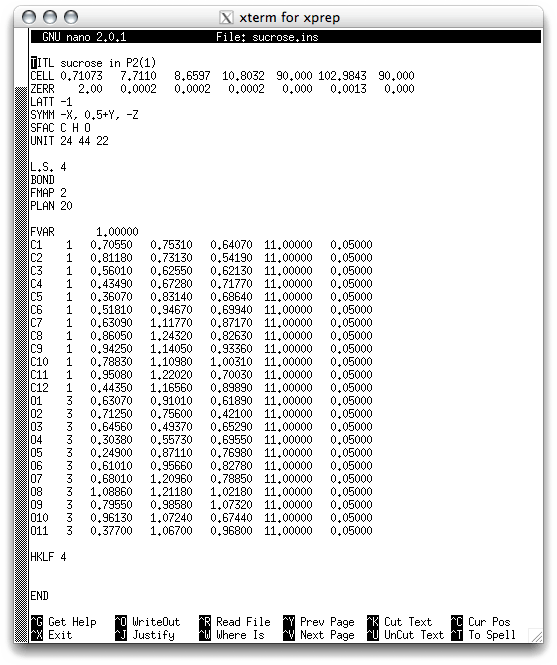
Some of the lines at the top should be familiar, and then there are a few new ones:
L.S. 4 - do four cycles of least-squares refinement (change number as needed).
BOND - calculate 'bond' distances and angles. (BOND $H include H atoms).
FMAP 2 - calculate a difference map ('2' = difference map).
PLAN 20 - find 20 peaks in the difference map (change number as needed).
FVAR - scale factor and special refinable parameters 'free variables'.
BOND - calculate 'bond' distances and angles. (BOND $H include H atoms).
FMAP 2 - calculate a difference map ('2' = difference map).
PLAN 20 - find 20 peaks in the difference map (change number as needed).
FVAR - scale factor and special refinable parameters 'free variables'.
The commands are explained in the SHELXL manual. At this stage it makes sense
to add a few additional lines to the sucrose.ins file. To do this, use a text
editor to make the following changes to the header, then save the changes and exit the
editor:

The meaning of some of these changes should be pretty obvious, while others may not
be so clear. One that you should be aware of is HTAB, which looks for
potential hydrogen bonds. You will encounter it again near the end of this part of
the tutorial. Feel free to consult the manual (always a good idea, and it is even
available on the web, try Google!). You may notice that the scattering factor number
for the oxygen atoms has been changed to 3. This was done by XP when you saved
the model with the file command.
To run SHELXL, simply type shelxl sucrose <return> at the terminal prompt. This will write a bunch of stuff to the screen, like this:
To run SHELXL, simply type shelxl sucrose <return> at the terminal prompt. This will write a bunch of stuff to the screen, like this:

This stuff is a brief summary of what happened during the SHELXL job. The
wR2 and R1 numbers are known as R-values, and measure how well
the model agrees with the experimental data. As the model improves, these numbers
decrease. Notice how wR2 drops from 57% to 20%. The output also tells us that
there are currently 2720 reflections in our dataset (but see later for the MERG
command), and 93 parameters (coordinates, displacement parameters, scale factor) in
the model. Down at the bottom it gives us a 'Highest peak' and 'Deepest hole'. These
are the largest features of the difference map, and are useful for deciding
how to further improve the model. The large peak here is 0.81 electrons per cubic
ångstrom, and it is 0.97Å from atom C4. This is more than likely a peak that
corresponds to a hydrogen atom on C4.
SHELXL has also written two files - a new sucrose.res, and a file called sucrose.lst. The new '.res' file contains the newly refined model, while the '.lst' file contains an extensive list of what went on during the SHELXL run. A couple of things should be apparent. First, the '.ins' file and the '.res' file share exactly the same format. SHELXL is fed a '.ins' file, and it writes out a '.res' file. The '.res' file is then edited (using XP or a text editor) to create a new '.ins' file, which is again refined using SHELXL. This process is repeated until no more improvements can be made. By the end of this part of the tutorial you will have a model that can't be improved (at least with the present dataset). The other thing that should be apparent is that SHELXL gives a lot of diagnostic information (the '.lst'. file). You won't need all the diagnostics for this tutorial because sucrose is quite straightforward, but with most structures you will likely have to consult the '.lst' file several times.
To see how the model has changed, you could look in the sucrose.res file. It should look like this:
SHELXL has also written two files - a new sucrose.res, and a file called sucrose.lst. The new '.res' file contains the newly refined model, while the '.lst' file contains an extensive list of what went on during the SHELXL run. A couple of things should be apparent. First, the '.ins' file and the '.res' file share exactly the same format. SHELXL is fed a '.ins' file, and it writes out a '.res' file. The '.res' file is then edited (using XP or a text editor) to create a new '.ins' file, which is again refined using SHELXL. This process is repeated until no more improvements can be made. By the end of this part of the tutorial you will have a model that can't be improved (at least with the present dataset). The other thing that should be apparent is that SHELXL gives a lot of diagnostic information (the '.lst'. file). You won't need all the diagnostics for this tutorial because sucrose is quite straightforward, but with most structures you will likely have to consult the '.lst' file several times.
To see how the model has changed, you could look in the sucrose.res file. It should look like this:

Notice that the '.res' file is getting pretty long now, even for this small structure,
so from now on only portions of the '.res' (or '.ins' etc.) files will be shown.
A few things are apparent from the file listing. Probably most obvious is the last
column of the atom lines. These numbers are all around 0.015 give or take a bit,
whereas they were all 0.05 before the SHELXL run. This confirms that SHELXL
really has changed the model. There are also small changes to the atom coordinates.
A new line with a suggested weighting scheme (WGHT) has been added, and all 20
of the requested peaks (PLAN 20) of the difference map (FMAP 2) have been
appended to the '.res' file as 'Q' peaks (the program uses 'Q' simply because there are
no elements with symbol Q). Let's take a look at the model and the difference map. It
should be possible to tell which of the Q peaks are hydrogen atoms.
To do this, type 'xp sucrose' at the terminal prompt. When the XP text window appears, type:
To do this, type 'xp sucrose' at the terminal prompt. When the XP text window appears, type:
fmol <return>
mpln/n <return>
proj <return>
mpln/n <return>
proj <return>
You should get this:

Some of these Q peaks look as though they might be tertiary C-H (e.g. Q1),
secondary C-H2 (e.g. Q9 & Q10), and even hydroxyl O-H
(e.g. Q15), but others look like nonsense (e.g. Q19, Q20). Also, even
some of the better looking Q peaks seem to make too many 'bonds'. The extra 'bond'
problem happens because XP interprets the Q peaks as carbon. You can improve
the picture a bit by telling XP that these Q peaks are H. To do this, EXIT the
XP graphical window (yellow button!), then in the XP text window type:
name q?? h?? <return>
name q? h? <return>
fmol <return>
proj <return>
name q? h? <return>
fmol <return>
proj <return>
The '?' acts as a wildcard character. You should now see this:

That looks a lot better, but notice there are still some H atoms missing (e.g.
at C11 and at O10), and the things close to O3 are no better now than they were when
they were called Q19 & Q20. At this point it you could keep the good looking
hydrogens in the model, remove the bad ones and refine again with SHELXL, but we
will not do that. Instead we will manually fix up the model with a text editor and add
the hydrogen atoms using a so-called riding model, which is usually a better
choice than free hydrogens (vide infra). Before we do that though, we
should check the position of the molecule in the unit cell using the cent
command. When you type cent <return> you should see something like this:

Since crystal structures are periodic in three dimensions, you can get molecular
fragments outside the 'unit cell box'. In other words, the centroid of a
molecular fragment may have coordinates that are <0 or >1. If that happens
you should consider shifting the structure (or part of the structure) to a
symmetry equivalent position so that its centroid coordinates are positive
numbers between 0 and 1. In the present case we don't need to shift the molecule
because its centroid is "within the box", but to illustrate the point let's move it
closer to the centre of the unit cell box.
In space group P21, the origin of the cell is arbitrary along b, which means it can be shifted anywhere along the b axis. Since our centroid has a b coordinate of 0.91377, if we move if by -0.41377 it will be exactly 0.5. For the a and c axes in P21 we don't have that much freedom (read the International Tables vol. A for details), but we can shift it by multiples of ±0.5. Since we want to get the centroid of the molecule closest to the middle of the cell box, we would leave a alone, shift b by -0.41377 and shift c by -0.5. To edit the '.res' file in this way, we need to get out of XP. Type exit <return> in the XP text window, but you may need to EXIT (yellow button) from the XP graphics window first.
Since we didn't file any changes made in XP, the '.res' previously written by SHELXL is still our most current model. Use a text editor to open the '.res' file, e.g. nano sucrose.res <return>, and add the following line to the '.res' file, on a new line below FVAR
In space group P21, the origin of the cell is arbitrary along b, which means it can be shifted anywhere along the b axis. Since our centroid has a b coordinate of 0.91377, if we move if by -0.41377 it will be exactly 0.5. For the a and c axes in P21 we don't have that much freedom (read the International Tables vol. A for details), but we can shift it by multiples of ±0.5. Since we want to get the centroid of the molecule closest to the middle of the cell box, we would leave a alone, shift b by -0.41377 and shift c by -0.5. To edit the '.res' file in this way, we need to get out of XP. Type exit <return> in the XP text window, but you may need to EXIT (yellow button) from the XP graphics window first.
Since we didn't file any changes made in XP, the '.res' previously written by SHELXL is still our most current model. Use a text editor to open the '.res' file, e.g. nano sucrose.res <return>, and add the following line to the '.res' file, on a new line below FVAR
MOVE 0 -0.41377 -0.5 1
This will shift the whole model within the unit cell box. The last number on this
line, 1, tells SHELXL to leave the handedness the same. If you need to
invert the model, this fourth number would be -1. Remember, with a
non-centrosymmetric structure there is a 50% chance that the initial model is
improperly inverted, no matter how good your data happen to be. By lucky chance we
have it correct in this example, but if you are feeling adventurous you could verify
this for yourself. Assignment of absolute structure using only the x-ray data
for light-atom crystals is quite specialized, and is beyond the scope of this tutorial.
Assigning absolute structure using information in x-ray data requires a measurable
difference (caused by anomalous dispersion) between Friedel pairs. Unfortunately
we simply don't get much anomalous signal using Mo Kα x-rays
(λ = 0.71073 Å) for light atom structures. In cases like this, it used to be
normal to merge the Friedel pairs using the SHELXL command MERG.
Nevertheless, you should know enough chemistry to assign the absolute configuration of
sucrose, and since you can ensure it is correct, you should ensure it
is correct.
Aside: Assignment of absolute structure is possible for light-atom structures with oxygen as the heaviest atom if you use Cu Kα x-rays (λ = 1.54178 Å), but even then extreme care is required. You also need to use a pair of instructions (TWIN & BASF) in the '.ins' file. That is beyond the scope of this tutorial, but you should be aware of the commands.
Back to the model at hand. Rather than add riding hydrogens now, we will instead make the heavy atoms anisotropic, using the ANIS command. You may have guessed by now that the order you do things is not fixed. Individual tastes and preferences vary, and even the type of structure can influence how you plan your refinement. In any event, the header for the '.res'. file should now look something like this:
Aside: Assignment of absolute structure is possible for light-atom structures with oxygen as the heaviest atom if you use Cu Kα x-rays (λ = 1.54178 Å), but even then extreme care is required. You also need to use a pair of instructions (TWIN & BASF) in the '.ins' file. That is beyond the scope of this tutorial, but you should be aware of the commands.
Back to the model at hand. Rather than add riding hydrogens now, we will instead make the heavy atoms anisotropic, using the ANIS command. You may have guessed by now that the order you do things is not fixed. Individual tastes and preferences vary, and even the type of structure can influence how you plan your refinement. In any event, the header for the '.res'. file should now look something like this:
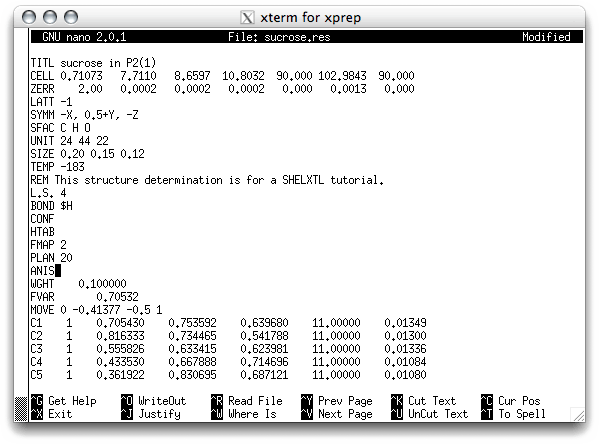
Save this as an '.ins' file.
To refine this new model with SHELXL, just type shelxl sucrose <return> at the terminal prompt. The SHELXL output should indicate that the R-values have dropped a bit. If you want to inspect the newly refined model with XP, go ahead, but it is not always necessary to use XP after every round of refinement. Instead, we will go straight to the new '.res' file for more manual editing. We're going to add riding hydrogen atoms to the carbons in the molecule, but we'll leave the hydroxyl H atoms for later. So edit the '.res' file and make the following changes:
To refine this new model with SHELXL, just type shelxl sucrose <return> at the terminal prompt. The SHELXL output should indicate that the R-values have dropped a bit. If you want to inspect the newly refined model with XP, go ahead, but it is not always necessary to use XP after every round of refinement. Instead, we will go straight to the new '.res' file for more manual editing. We're going to add riding hydrogen atoms to the carbons in the molecule, but we'll leave the hydroxyl H atoms for later. So edit the '.res' file and make the following changes:

You may also notice something different about the atom lines above. With anisotropic
displacement parameters ('ellipsoids'), there is too much stuff to fit on one
line, so they continue on the line below. Continuation lines like this are indicated
by the '=' sign.
The SHELXL command for adding riding hydrogen atoms is HFIX. This command uses numerical codes to specify the type of hydrogen to be added. In sucrose there are tertiary R3CH (HFIX 13), secondary R2CH2 (HFIX 23), and hydroxyl OH (HFIX 83 or HFIX 147) hydrogens. Other common types include methyl groups (HFIX 137 or HFIX 33) and aromatic/vinylic type (HFIX 43), but these are not present in sucrose. See the manual for full details of riding models. If you typed the HFIX instructions suggested above, SHELXL will add 14 hydrogen atoms to your model. The term riding model means that the hydrogen atoms ride on the heavy atom to which they are bonded. The riding model concept is very useful because it enables the hydrogens to be added easily, but without adding lots of additional parameters. It is worth pointing out here that SHELXL allows several ways to tweak these riding models. Such fine points are beyond this tutorial, but you should be aware that additional options exist. Consult the manual for more information.
Some H atoms are unambiguous, e.g. in R3CH and R2CH2, the H are fixed relative to the adjacent atoms. Others are not so easily positioned e.g. hydroxyl (and methyl), because they have torsional freedom. When adding hydrogens, it is best to first find evidence in a difference map, and then add them with HFIX. It is good practice to start with the unambiguous hydrogens, and add the less well-defined hydrogens later. SHELXL has a few tricks to locate these sorts of problem hydrogens, and we will employ one such trick for the eight remaining OH hydrogen atoms soon.
Anyhow, save it as a new '.ins' file, and run SHELXL (you should be able to do this on your own by now). As always, SHELXL writes stuff to the screen.
The SHELXL command for adding riding hydrogen atoms is HFIX. This command uses numerical codes to specify the type of hydrogen to be added. In sucrose there are tertiary R3CH (HFIX 13), secondary R2CH2 (HFIX 23), and hydroxyl OH (HFIX 83 or HFIX 147) hydrogens. Other common types include methyl groups (HFIX 137 or HFIX 33) and aromatic/vinylic type (HFIX 43), but these are not present in sucrose. See the manual for full details of riding models. If you typed the HFIX instructions suggested above, SHELXL will add 14 hydrogen atoms to your model. The term riding model means that the hydrogen atoms ride on the heavy atom to which they are bonded. The riding model concept is very useful because it enables the hydrogens to be added easily, but without adding lots of additional parameters. It is worth pointing out here that SHELXL allows several ways to tweak these riding models. Such fine points are beyond this tutorial, but you should be aware that additional options exist. Consult the manual for more information.
Some H atoms are unambiguous, e.g. in R3CH and R2CH2, the H are fixed relative to the adjacent atoms. Others are not so easily positioned e.g. hydroxyl (and methyl), because they have torsional freedom. When adding hydrogens, it is best to first find evidence in a difference map, and then add them with HFIX. It is good practice to start with the unambiguous hydrogens, and add the less well-defined hydrogens later. SHELXL has a few tricks to locate these sorts of problem hydrogens, and we will employ one such trick for the eight remaining OH hydrogen atoms soon.
Anyhow, save it as a new '.ins' file, and run SHELXL (you should be able to do this on your own by now). As always, SHELXL writes stuff to the screen.
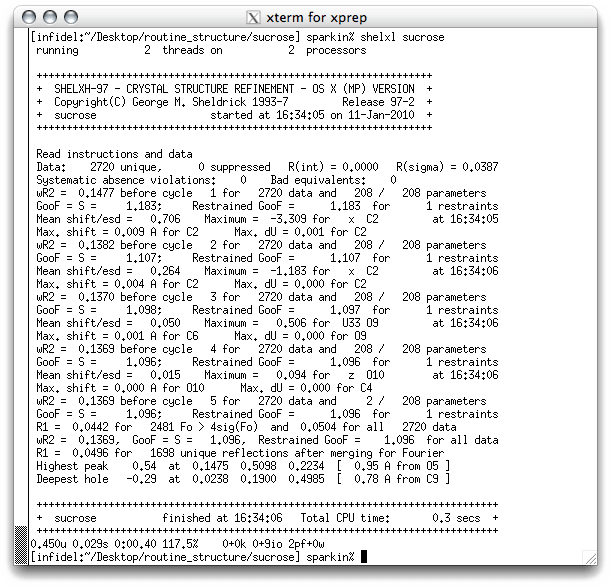
Notice the R-values are a lot lower, and the difference map
features are smaller. If you open the '.res' file with a text editor you can see how
SHELXL has included the hydrogen atoms:

For the riding hydrogens added to carbon, SHELXL has added AFIX
lines with the same code as the HFIX instruction used to generate them. It also
ends each grouping with AFIX 0. The H atoms are numbered according to their
parent atom, and they have a '2' in the scattering factor column (H is the
second element on the SFAC line). The H atoms are given an occupancy of 11.00000
(i.e. fixed at unit occupancy), and a thermal (displacement) parameter,
Uiso, of -1.2. This special form of the thermal parameter tells SHELXL
to assign an isotropic thermal parameter to the H atom that is 20% larger than
the equivalent isotropic thermal parameter of the parent atom. The value of -1.2
is usual for H atoms whose position is fixed relative to the parent atom. Other types
of hydrogen (e.g. methyl and hydroxyl) have torsional freedom, and for these a
value of -1.5 (i.e. 50% larger than the parent atom) is more appropriate.
It usually helps to look at the heights of difference map peaks. As stated above, the difference map peaks are appended to the '.res' file so you can see them with a text editor, but we can also list them within XP. To do this, start XP by typing xp sucrose <return>.
As always, the first thing to enter in the XP text window is fmol <return>. To see the difference map peak heights, type info <return>, which gives you this:
It usually helps to look at the heights of difference map peaks. As stated above, the difference map peaks are appended to the '.res' file so you can see them with a text editor, but we can also list them within XP. To do this, start XP by typing xp sucrose <return>.
As always, the first thing to enter in the XP text window is fmol <return>. To see the difference map peak heights, type info <return>, which gives you this:

Remember that we have 8 hydroxyl hydrogens to add. The new Q peaks are all pretty small
(last column - the units are eÅ-3), but there is a sort of step from Q8 to Q9,
so it is likely that the top most peaks correspond to hydrogen atoms. The best way to
tell is to view a projection (proj <return>).

Argh! That looks kind of nasty. To simplify and improve the view, you need to remove
the Q peaks that are incorrect (kill or pick), edit the Q peaks that are
good to be H (pick or name), and issue another fmol command. Once
this is done you should have something like this:

You could save this model (file sucrose <return> etc.) and continue
to work with the OH hydrogens as they are. If you did this, the hydroxyl H atoms would
refine freely (i.e. they would not ride). For x-ray data, however, it is
usually (but not always!) better to use a riding model, as you did earlier with
the H atoms on carbon. This is what we will do here so that you learn the ways
SHELXL can handle riding models for OH groups.
For hydroxyl groups there are a couple of options. If the data are high quality (i.e. low temperature, good crystal), and you can find promising difference map peaks, then the riding model to try first uses the command HFIX 147. This tells SHELXL to compute the electron density in a toroid (donut) out beyond the oxygen in question, and place the H atom at the position of maximum electron density. If HFIX 147 generates an impossible or improbable position for any of the H atoms then try the next best option, HFIX 83. This variant searches for plausible hydrogen bond acceptors to place the H atom. Since the sucrose data used in this tutorial are good, we'll try HFIX 147 first. With good data, HFIX 147 and HFIX 83 usually add H atoms at about the same place.
Edit the '.res' file from the previous SHELXL run, and add the following line:
For hydroxyl groups there are a couple of options. If the data are high quality (i.e. low temperature, good crystal), and you can find promising difference map peaks, then the riding model to try first uses the command HFIX 147. This tells SHELXL to compute the electron density in a toroid (donut) out beyond the oxygen in question, and place the H atom at the position of maximum electron density. If HFIX 147 generates an impossible or improbable position for any of the H atoms then try the next best option, HFIX 83. This variant searches for plausible hydrogen bond acceptors to place the H atom. Since the sucrose data used in this tutorial are good, we'll try HFIX 147 first. With good data, HFIX 147 and HFIX 83 usually add H atoms at about the same place.
Edit the '.res' file from the previous SHELXL run, and add the following line:
HFIX 147 O2 O3 O4 O5 O8 O9 O10 O11
Save the file as a new '.ins' file, and refine it with SHELXL. By this point in
the tutorial, you should be able to do this, and read the new '.res' file into XP
on your own. From the SHELXL output, a few things are apparent. The R-values
are much lower and the difference map is very flat. The mean and max
shifts are also small, which indicates that the refinement is converging well.
Convergence is a necessary criterion for a finished structure.
With luck the 8 new riding H atoms will be chemically sensible, but don't assume they're ok - you have to check them. In XP, the structure should look like this if you remove all the Q peaks (use either kill $Q or fmol less $Q):
With luck the 8 new riding H atoms will be chemically sensible, but don't assume they're ok - you have to check them. In XP, the structure should look like this if you remove all the Q peaks (use either kill $Q or fmol less $Q):

It looks good! The model is essentially complete, but it is not quite finished. There
are a few more things that need to be done before this refinement can be considered
complete. The first concerns the weighting scheme, which is given on the WGHT
line in the '.res' (or '.ins') file. If you edit the '.res' file you will see that the
WGHT line still has its default parameter, 0.1000. There are a number of ways to
devise weighting schemes, and there are strong scientific arguments for and against
different schemes. The accepted weights for use in SHELXL are optimized so as to
give a goodness-of-fit (Goof) as close to 1.00 as possible. Once all the atoms have been
found it is time to start adjusting the weights. Whether or not the SHELXL weights
are actually the best is another matter entirely. Such philosophical minutiae
are beyond the scope of this tutorial, so we will just follow convention.
Further down the '.res' file, SHELXL suggests suitable weights for the next round of refinement. All you have to do is copy this suggested WGHT line and replace the old one in the '.res' file. Optimizing the weights in this way often requires a few iterations. By now you should be able to update the WGHT and refine with SHELXL a few times, and the tutorial assumes you have done this. You should find that the R-values are a bit smaller, the difference map is a bit flatter, and the shifts are very small.
Another tweak, that may or may not be needed, is an extinction correction. This should only be tested once the model is essentially complete, so now is a good time to try it. You should know that the physics of extinction is pretty complex. It tends to affect only the strongest reflections at low diffraction angle, and it manifests itself as a reduction (extinction) of the affected reflections. Suffice to say, the correction available in SHELXL is only an approximation. You can get some idea of when an extinction correction will help by comparing observed and calculated intensities (or F2 values). This is where the '.lst' file comes in! Somewhere in the '.lst' file there is a table like this, and you need to find it:
Further down the '.res' file, SHELXL suggests suitable weights for the next round of refinement. All you have to do is copy this suggested WGHT line and replace the old one in the '.res' file. Optimizing the weights in this way often requires a few iterations. By now you should be able to update the WGHT and refine with SHELXL a few times, and the tutorial assumes you have done this. You should find that the R-values are a bit smaller, the difference map is a bit flatter, and the shifts are very small.
Another tweak, that may or may not be needed, is an extinction correction. This should only be tested once the model is essentially complete, so now is a good time to try it. You should know that the physics of extinction is pretty complex. It tends to affect only the strongest reflections at low diffraction angle, and it manifests itself as a reduction (extinction) of the affected reflections. Suffice to say, the correction available in SHELXL is only an approximation. You can get some idea of when an extinction correction will help by comparing observed and calculated intensities (or F2 values). This is where the '.lst' file comes in! Somewhere in the '.lst' file there is a table like this, and you need to find it:

Notice how the observed Fo^2 are systematically smaller than the calculated Fc^2 for
those reflections with large values of Fo^2 or Fc^2, and that these happen to have the
larger numbers in the resolution column. This may be caused by extinction (but
may also be due to overloaded intensities or to reflections hiding behind the beamstop.
Both of those possibilities need to be investigated before worrying about extinction.
To apply a correction, edit the '.res' file and add a statement EXTI above the
atom lines, re-save it as an '.ins', and refine with SHELXL.
If you did it properly, the SHELXL output should look like this:
If you did it properly, the SHELXL output should look like this:

The SHELXL output tells you that the addition of EXTI was probably a good
thing. You can tell this because in the first cycle, the shift/esd of the EXTI
parameter was >5, and in the second cycle it was >2, i.e. the EXTI
parameter is much larger than its uncertainty. Generally speaking, EXTI
should be more than about 3 times its uncertainty (aka su or esd) for you
to consider it valid. The definitive check is again given in the .lst file, which
lists the EXTI parameter and its esd (estimated standard deviation)
after each least-squares cycle. For this example we have:

From the above picture, you should be able to figure out that the EXTI parameter
is about 7 times its esd. At this stage the model is pretty much finished, but
it may be worth changing the weights (WGHT) one more time. Go ahead and do it,
and don't forget to refine with SHELXL.
Even though the model is now refined to completion, we are not quite done. Although we have all the information necessary to describe the structure, we do not yet have it in the format required for journal and database deposition. That's right, we need the dreaded CIF (crystallographic information file) format. Luckily, SHELXL will write a CIF if there is an ACTA statement in the '.ins' file. Go ahead and add this and run SHELXL.
With a structure like sucrose, there are lots of hydrogen bonds that should be in the CIF. To add these properly requires a bit of tedious manual editing of the '.ins'. Remember near the beginning of this tutorial you added a command HTAB ? Now you're going to use it the ensure that H-bond information is added to the CIF.
The simple form of HTAB you added earlier tells SHELXL to analyze the structure model for hydrogen bonds. If it finds any then it writes these near the end of the '.lst' file. The list looks like this:
Even though the model is now refined to completion, we are not quite done. Although we have all the information necessary to describe the structure, we do not yet have it in the format required for journal and database deposition. That's right, we need the dreaded CIF (crystallographic information file) format. Luckily, SHELXL will write a CIF if there is an ACTA statement in the '.ins' file. Go ahead and add this and run SHELXL.
With a structure like sucrose, there are lots of hydrogen bonds that should be in the CIF. To add these properly requires a bit of tedious manual editing of the '.ins'. Remember near the beginning of this tutorial you added a command HTAB ? Now you're going to use it the ensure that H-bond information is added to the CIF.
The simple form of HTAB you added earlier tells SHELXL to analyze the structure model for hydrogen bonds. If it finds any then it writes these near the end of the '.lst' file. The list looks like this:

This tells you which atoms are involved in hydrogen bonds as donors and acceptors. Some
of the acceptors may happen to be on symmetry equivalent molecules, and the program
tells you the symmetry operation involved. To get this stuff into the CIF, you need to
enter it in a '.ins' file for a final pass through SHELXL, but it needs a bit of
re-formatting to get the syntax right. Since our best model is in the previous '.res'
file, we'll put the special HTAB instructions in there and re-save as a '.ins.
The edited '.ins' file should look like this

Hydrogen bonding in sucrose is quite extensive, so there's a lot of them to specify.
The equivalent positions are each put on separate EQIV instructions, and
each H-bond gets its own HTAB command. In sucrose, most of the H-bonds are
between symmetry related molecules, but two of them are between atoms in the same
asymmetric unit. These are the ones that don't have one of the equivalent positions
appended. You could also increase the number of least-squares cycles (e.g.
L.S. 6) to ensure convergence.
To complete this part of the sucrose tutorial, save this as a new '.ins' file, and run it through SHELXL once or twice. The refinement is now finished, and you are ready to draw structure diagrams and to edit and validate the CIF.
To complete this part of the sucrose tutorial, save this as a new '.ins' file, and run it through SHELXL once or twice. The refinement is now finished, and you are ready to draw structure diagrams and to edit and validate the CIF.
Part 1: Setting up instructions - XPREP
Part 2: Structure solution - SHELXS
Part 3: Molecule editing - XP
Part 4: Structure refinement - SHELXL
Part 5: Ellipsoid and packing plots - XP
Part 6: The Crystallographic Information File - CIF
Part 2: Structure solution - SHELXS
Part 3: Molecule editing - XP
Part 4: Structure refinement - SHELXL
Part 5: Ellipsoid and packing plots - XP
Part 6: The Crystallographic Information File - CIF
Return to the first page of this tutorial
Return to the main Tutorials page or to the main X-Ray Lab page
Return to the main Tutorials page or to the main X-Ray Lab page