Absorption correction using SADABS with KappaCCD data
Note: This part of the tutorial is out of date, and will not work properly with the newest versions of SADABS. The newest versions of SADABS require the use of the utility program 'x2sad', that converts the information in the Denzo .x files to a form readable by SADABS. There is a clickable pull-down menu item in the SuperGUI window under "Alternatives" that will run x2sad for you and create a file named "denzo.sad" for use with SADABS.
This is a quick run-through showing the steps involved in using SADABS to correct for anisotropic absorption with data from the kappaCCD diffractometer.
In order to prepare the data for SADABS correction it should be processed using scalepack similar to step 2 in the data reduction scheme, but we need to suppress the merging and scaling of the data. You have seen the following window before ...
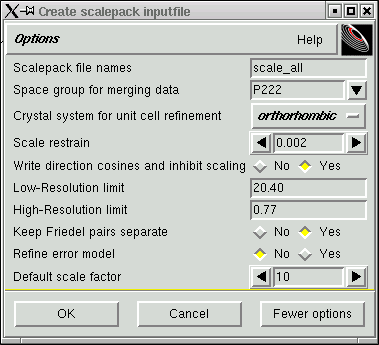
... notice now, however, that "yes" is selected on the "Write direction cosines ..." line. The "direction cosines" referred to here define the direction of the incident and diffracted x-ray beams for each reflection in the dataset. When you are satisfied, click "OK" and you get this:
You have seen this before also. If you need to reject a particular scan (very rare), do it here then click "OK". A small yellow window will appear briefly, but it will soon disappear - scalepack runs much faster when it inhibits the scaling process. When it is done the following window appears:
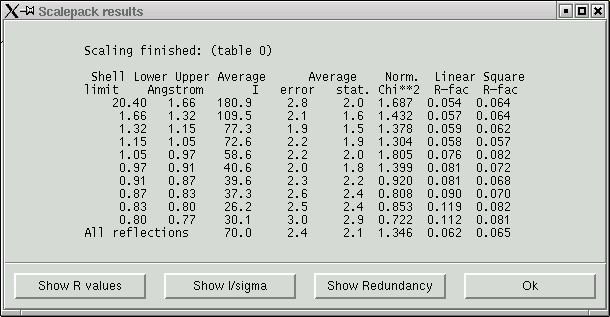
Again, this is just like the scalepack statistics near the end of section 2 of this data processing guide. When you click "OK" the program will go ahead and write a file named "scale_all.sca" with all the data and all the direction cosines. Unfortunately SADABS cannot read this file directly so we need to re-format it using the "cifin" utility (recall that cifin was also used in section 3 of this guide). Invoke cifin from the terminal shell window ...
... and you get a dialog window like this:
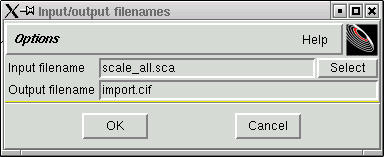
Change the output filename as per the following suggestion:
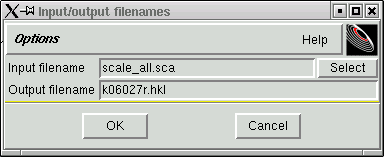
Notice that this new filename has the standard form of filenames used with the kappaCCD machine, followed by an "r", which stands for "raw", and there is the extension ".hkl". SADABS will read this raw file. When you click "OK" the data are reformatted and the progress of this reformatting process is printed out in the terminal window like this:
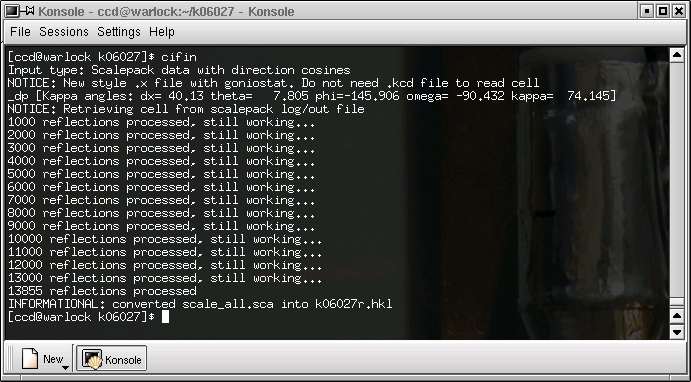
It is now time to run SADABS, so do the obvious thing - type "sadabs" followed by the return key on the command line of the terminal window ...
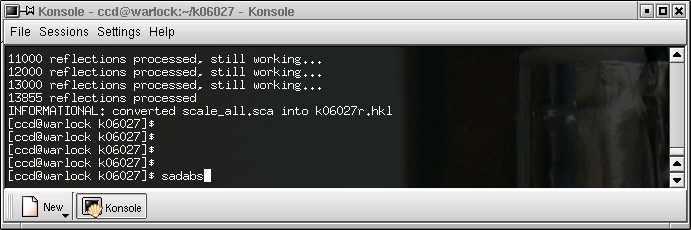
... and you will get something like this:
The program initially suggets a listing filename of "sad.abs", but it makes more sense to keep all your files with the same naming convention, as above.
The next step is to inform SADABS of the Laue group:
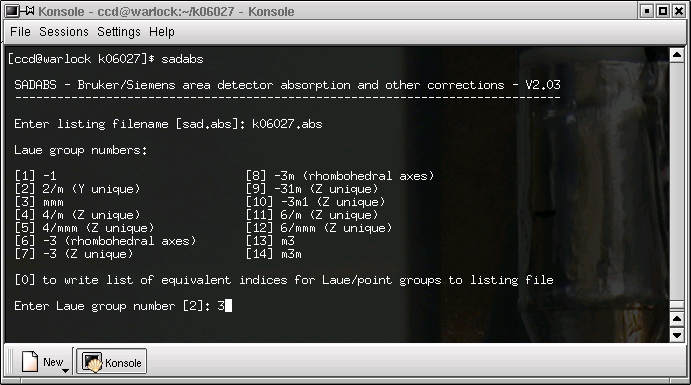
In this case the crystal was orthorhombic so the Laue group has to be mmm, so we change from the default value.
For the most part the defaults are sensible in this program so this is largely a matter of pushing the return key a bunch of times.
Enter the name of your "raw" datafile - the one with direction cosines that you made earlier.
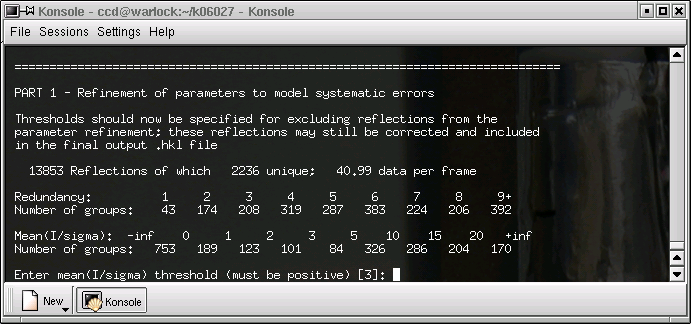
The defaults are usually ok, hit return ...
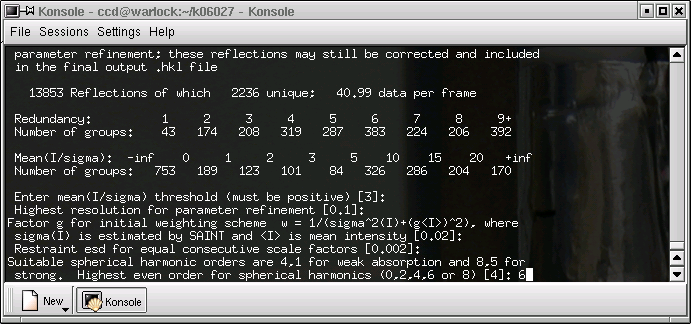
Here it sometimes pays to change the defaults. If you expect the absorption to be severe, increase this value. Be aware that it is possible to be over zealous, and that is a bad thing.
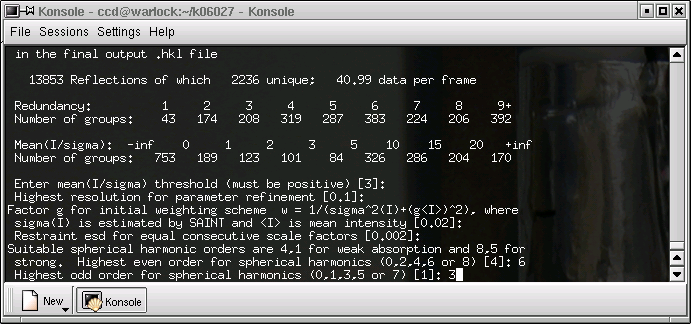
Again, change the value if you know what you are doing. It is worth doing some experimentation here to find the best value.
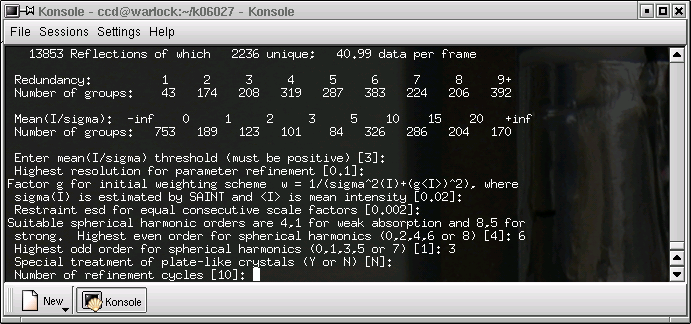
The defaults are probably ok, so hit return. Sometimes it helps to increase the number of cycles to (say) 20.
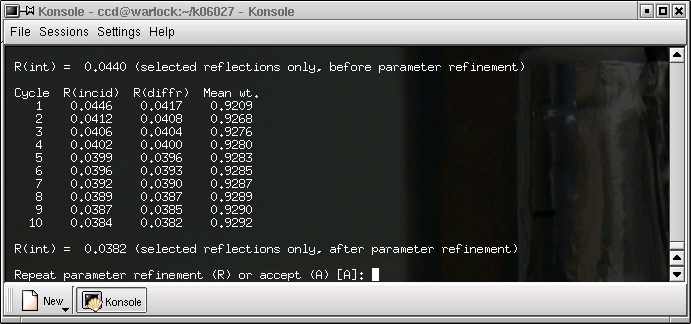
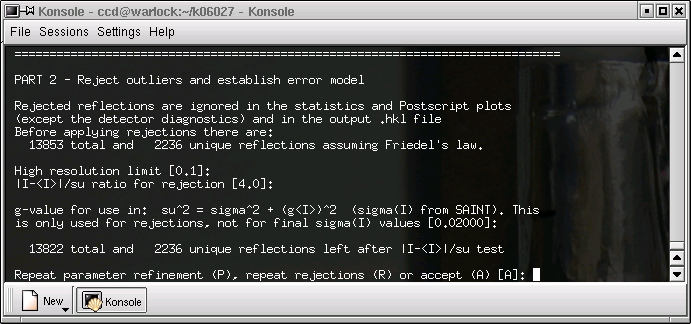
SADABS shows the progress of its refinement of the parameters that it uses to define the absorption correction. The R(int) value should get lower and it should be approximately the same for the incident and diffracted x-rays. If the refinement is accepted, we get this:
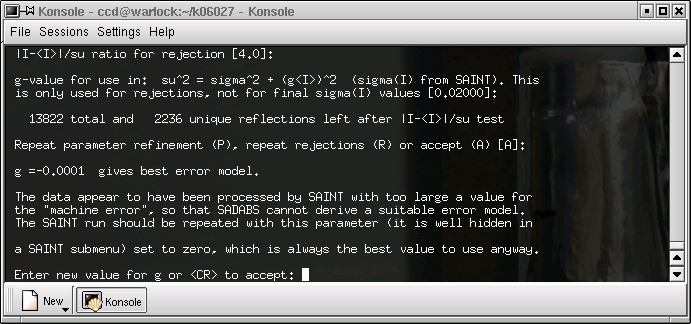
It is unlikely that anything will be changed here, so just hit return ...
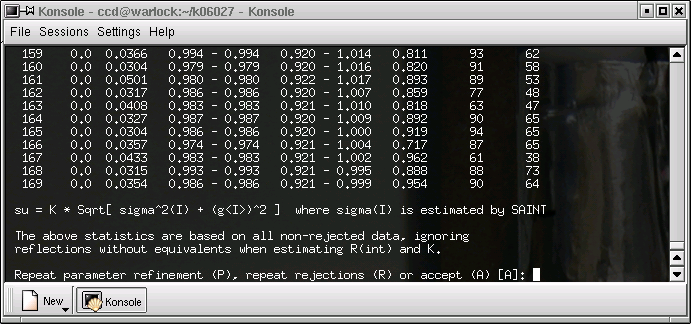
...and again ...
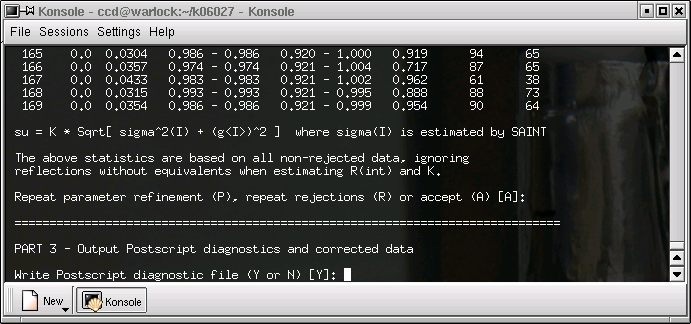
... and again ...
... and again ...
... and again ...
... and again ...
... and again ...
... and again (but feel free to rename the file if you like) ...
... and again ...
... and again ...
... here you will probably want to use a filename that corresponds to your sequence number. In the example above, the "s" in the filename indicates that this file has been run through SADABS.
The main function of SADABS is to correct for the anisotropic (3 spatial directions) part of the absorption effects. In reality there is also a fourth component caused by the fact that absorption changes as a function of resolution. You have the option of correcting for this as well, but here it will be ignored - hit return.
Normally just hit return ...
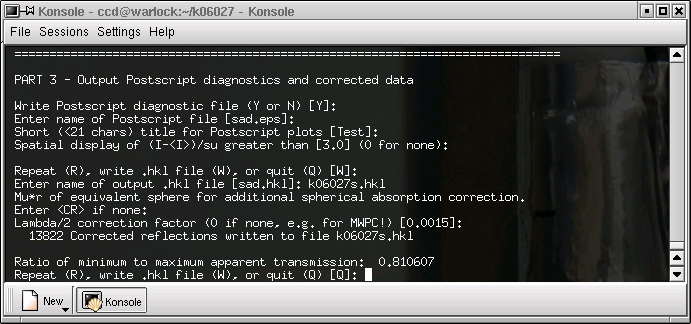
... and finally the program ends. Make a note of the minimum to maximum transmission coefficient ratio. This number will be used in the final edit of your complete CIF after the structure is solved and refined.
Go back to the main Kappa data processing page
Return to the main Tutorials page or to the main X-Ray Lab page