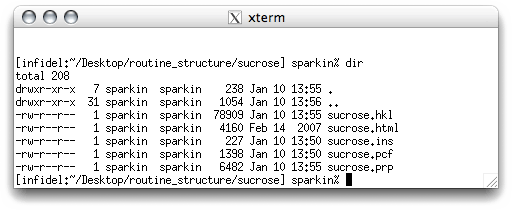
The file sucrose.ins has instructions to solve the structure. If you remember,
this needs to be edited to correct the values of the uncertainties of the cell
parameters. To edit this file you need to use a text editor. Whatever you do do
not use a word processor for this, ever. On linux there are several text
editors available (vi, pico, nano, emacs etc.).
These are also available on the Mac (as are BBEdit, TextWrangler,
etc.). If you find yourself restricted to Microsoft's Windows, other options
are available (e.g. WordPad, NotePad). Just remember to never
use MS Word because it is not a text editor!
In this example we will use nano for editing text. At the unix prompt, type 'nano sucrose.ins', and hit <return>.
In this example we will use nano for editing text. At the unix prompt, type 'nano sucrose.ins', and hit <return>.
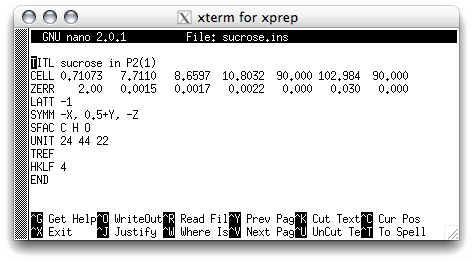
Change the uncertainties on the ZERR line, as follows:
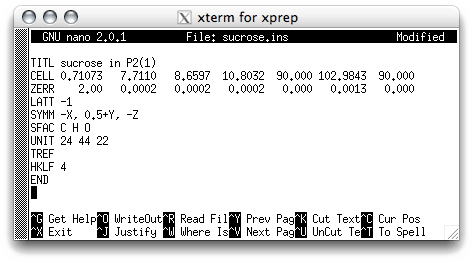
In this case there is an extra significant figure in the beta cell angle, so you should
edit that as well. Remember, the proper values to use are in the nreport.html
file. In the '.ins' file, all instructions and commands are four characters long, and
some have additional information. Full descriptions are in the SHELXTL manual,
but briefly:
TITL - contains the title. You can add stuff here if you like.
CELL - x-ray wavelength followed by the unit cell dimensions.
ZERR - 'Z', the number of formula units, followed by the unit cell uncertainties.
LATT - centering conditions, centrosymmetric or non centrosymmetric.
SYMM - crystallographic symmetry, here it's a 21 screw axis.
SFAC - scattering factors to use (C = carbon, H = hydrogen, O = oxygen etc.)
UNIT - how many of each type of atom are in the unit cell.
TREF - Tangent REFinement, this is what solves the structure.
HKLF - reads in the data. '4' specifies the format of the data.
END - end of instructions used by SHELXS.
CELL - x-ray wavelength followed by the unit cell dimensions.
ZERR - 'Z', the number of formula units, followed by the unit cell uncertainties.
LATT - centering conditions, centrosymmetric or non centrosymmetric.
SYMM - crystallographic symmetry, here it's a 21 screw axis.
SFAC - scattering factors to use (C = carbon, H = hydrogen, O = oxygen etc.)
UNIT - how many of each type of atom are in the unit cell.
TREF - Tangent REFinement, this is what solves the structure.
HKLF - reads in the data. '4' specifies the format of the data.
END - end of instructions used by SHELXS.
Later you'll see that there can be other stuff after this END. Type <control>X
to get out of the nano editor. It will ask you if you want to save the changes.
You should be able to figure this all out, it is quite straightforward.
To solve the structure using SHELXS, simply type 'shelxs sucrose' at the prompt, and hit <return>. This is what happens:
To solve the structure using SHELXS, simply type 'shelxs sucrose' at the prompt, and hit <return>. This is what happens:

To a trained eye it is clear from this output that the structure is solved. Successful
solutions are indicated by small CFOM, NQUAL values close to -1, and RE values less
than about 0.25. As you gain experience you will be able to interpret this output to
diagnose problems (not all structures solve so easily!). If you now do a directory
listing you will see two new files: sucrose.lst and sucrose.res. The
'.lst' file is a listing of what happened during the SHELXS run. It contains a
wealth of useful information, but much of it is too specialized to worry about yet.
The file we are interested in is the '.res' file, which contains results.
Remember, '.ins' = instructions, '.res' = results.
You could look at the '.res' file with a text editor (e.g. nano) if you like, but it probably won't mean much yet. If you choose to look, it should look like this:
You could look at the '.res' file with a text editor (e.g. nano) if you like, but it probably won't mean much yet. If you choose to look, it should look like this:
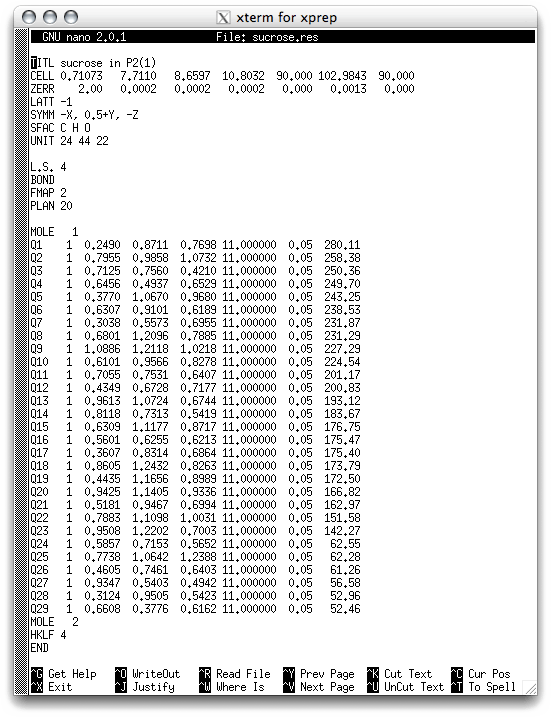
You may recognize some of the lines in the header from the old sucrose.ins file.
There are also some new lines, which will be explained in detail later. The important
parts as far as the structure model is concerned are the lines that begin with Q.
These are the putative atomic coordinates of the initial structure solution. These
lines have eight columns:
Column 1: These are atom labels, and will be changed as necessary.
Column 2: The position on the SFAC line of the atom in question, again change as necessary.
Columns 3,4,5: Atom coordinates, given as fractions of unit cell edges.
Column 6: Occupancy factor of the atom. In SHELX, variables are fixed by adding 10 to, so '11.000000' is interpreted as "occupancy fixed at 1.000000". This may seem confusing at first, but it is easy to get used to. Atoms are most often present at full occupancy, but not always (e.g. disordered atoms).
Column 7: This number (aka Uiso) is the mean-square displacement amplitude of the atom. The default value of 0.05 is typical for room temperature work, and so is probably too high for this structure. These numbers will change as the structure model is improved by refinement. Later, when you refine the structure, this single value (think of it as a radius that describes a sphere) will be replaced by six numbers (these define an ellipsoid - anisotropic displacement parameters) as the refinement nears completion. You can get an estimate of the r.m.s. amplitude of atomic vibration by taking the square root of this Uiso.
Column 8: The height of the peak in the structure solution E-map for the atom. Larger values correspond to 'bigger' atoms. Notice that there is a steady decrease down to Q23, and then a big jump down for Q24. To a trained eye it is likely that Q24-Q29 are not real atoms, just noise peaks in the initial model.
Column 2: The position on the SFAC line of the atom in question, again change as necessary.
Columns 3,4,5: Atom coordinates, given as fractions of unit cell edges.
Column 6: Occupancy factor of the atom. In SHELX, variables are fixed by adding 10 to, so '11.000000' is interpreted as "occupancy fixed at 1.000000". This may seem confusing at first, but it is easy to get used to. Atoms are most often present at full occupancy, but not always (e.g. disordered atoms).
Column 7: This number (aka Uiso) is the mean-square displacement amplitude of the atom. The default value of 0.05 is typical for room temperature work, and so is probably too high for this structure. These numbers will change as the structure model is improved by refinement. Later, when you refine the structure, this single value (think of it as a radius that describes a sphere) will be replaced by six numbers (these define an ellipsoid - anisotropic displacement parameters) as the refinement nears completion. You can get an estimate of the r.m.s. amplitude of atomic vibration by taking the square root of this Uiso.
Column 8: The height of the peak in the structure solution E-map for the atom. Larger values correspond to 'bigger' atoms. Notice that there is a steady decrease down to Q23, and then a big jump down for Q24. To a trained eye it is likely that Q24-Q29 are not real atoms, just noise peaks in the initial model.
Ok, reading text files is pretty dull. What does this actually look like ? We
need to view the structure to decide how to proceed. For this, SHELXTL has a
very powerful molecular editor called XP. Note that the XP in SHELXTL
predates Microsoft's XP by decades, and is considerably better at its job. We
will use XP for editing the initial structure model in the third part of this
tutorial.
Part 1: Setting up instructions - XPREP
Part 2: Structure solution - SHELXS
Part 3: Molecule editing - XP
Part 4: Structure refinement - SHELXL
Part 5: Ellipsoid and packing plots - XP
Part 6: The Crystallographic Information File - CIF
Part 2: Structure solution - SHELXS
Part 3: Molecule editing - XP
Part 4: Structure refinement - SHELXL
Part 5: Ellipsoid and packing plots - XP
Part 6: The Crystallographic Information File - CIF
Return to the first page of this tutorial
Return to the main Tutorials page or to the main X-Ray Lab page
Return to the main Tutorials page or to the main X-Ray Lab page