
All that remains to be done is to assign the major and minor components to
separate PARTs, add hydrogen atoms, and refine anisotropic displacement
parameters for the major component. For the minor disorder component, there is
little justification for going beyond the isotropic level because the occupancy
fraction is so small. (It can be done, but requires special treatment - see
section 5 if you are curious.)
To proceed, we'll edit the *.res file to insert the required PARTs, make the major component anisotropic, and throw in a few restraints to ensure that the model continues to make chemical sense throughout subsequent refinement runs.
To proceed, we'll edit the *.res file to insert the required PARTs, make the major component anisotropic, and throw in a few restraints to ensure that the model continues to make chemical sense throughout subsequent refinement runs.
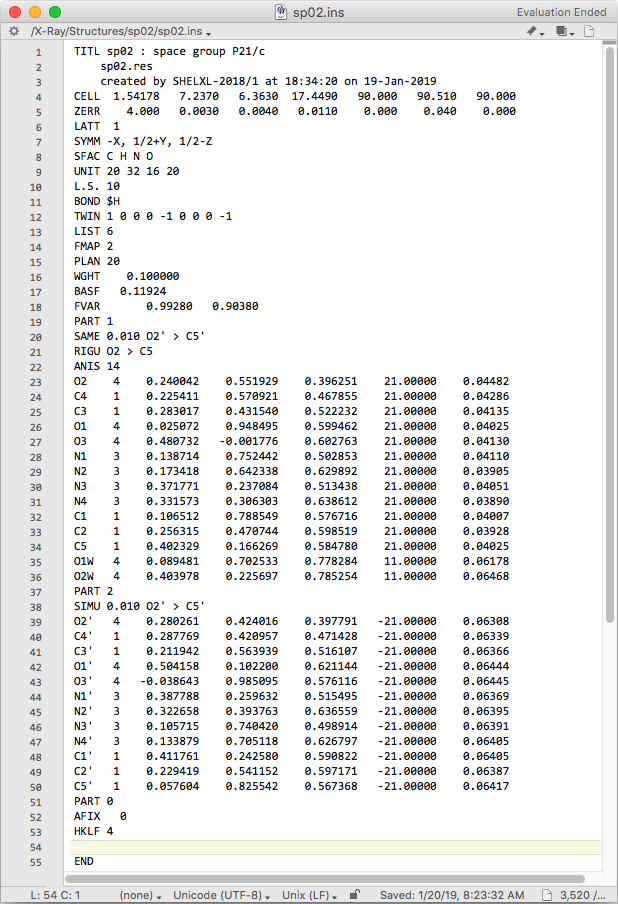
In the above file, the SIMU restraints have been removed for the major
component and relaxed considerably for the minor. PART numbers have been
assigned to both uric acid fragments, a SAME restraint has been added,
which should ensure that the minor component geometry closely matches that of
of the much better defined major component. Finally, the major component is set
to gain anisotropic displacement parameters. The RIGU restraint here is
just a precaution: it may or may not be needed, but it does no harm. As a later
exercise, you could try removing this RIGU to see what happens. The minor
component displacement parameters are still constrained to be isotropic. When
refined with SHELXL, the result looks like this:

The refinement is looking good. The largest difference map peaks look to be
consistent with hydrogen atom positions on the uric acid nitrogen atoms and on
the water molecules. Since their geometry is well known, ShelXle should
be able to add the N-H hydrogens automatically, or you could add them using
HFIX 43. Since major and minor PARTs have slightly different
hydrogen atom positions, it would be unwise to try to refine these hydrogens
freely. There is no convenient way to automatically add the water hydrogens, so
they will have to be assigned using difference map peaks. To be on the safe
side, we'll add the uric acid hydrogens first, refine, then add the water hydrogens,
perhaps with O-H distance and H-O-H 'angle-distance' restraints. Not shown here,
the water molecules should be set to PART 0, as they are not disordered.
Once all the atoms are included, the weighting scheme can be optimized and other
minor things such as testing for extinction can be performed. The end result
after all these tasks should look something like this:

The refinement should now satisfy even the most pedantic referee. The final
R-value is 3.78%, the difference map is essentially flat (largest features
are +0.167 and -0.137 eÅ-3), the weighting scheme looks normal,
etc. All that remains is to create and edit the CIF. Having said that, it
is possible to make a few tweaks to the model that will reduce the number
of parameters needed, albeit by adding a substantial degree of complexity. See
below for section 5 if you are interested.
1) Solve structure using SHELXT and ShelXle.
2) Include twinning using TWIN and BASF in SHELXL.
3) Add disorder using FRAG...FEND in SHELXL.
4) Add restraints/constraints to enable stable refinement.
... More advanced stuff (only possible because β is very close to 90°)...
5) Tie the major/minor disorder components using FVARs.
2) Include twinning using TWIN and BASF in SHELXL.
3) Add disorder using FRAG...FEND in SHELXL.
4) Add restraints/constraints to enable stable refinement.
... More advanced stuff (only possible because β is very close to 90°)...
5) Tie the major/minor disorder components using FVARs.
Return to the first page of this tutorial
Return to the main Tutorials page or to the main X-Ray Lab page
Return to the main Tutorials page or to the main X-Ray Lab page