6/7: Nine tin complexes
For this example we'll superimpose nine closely related tin complexes from a recent paper in the Journal of Molecular Structure.
Here's what the structures look like:
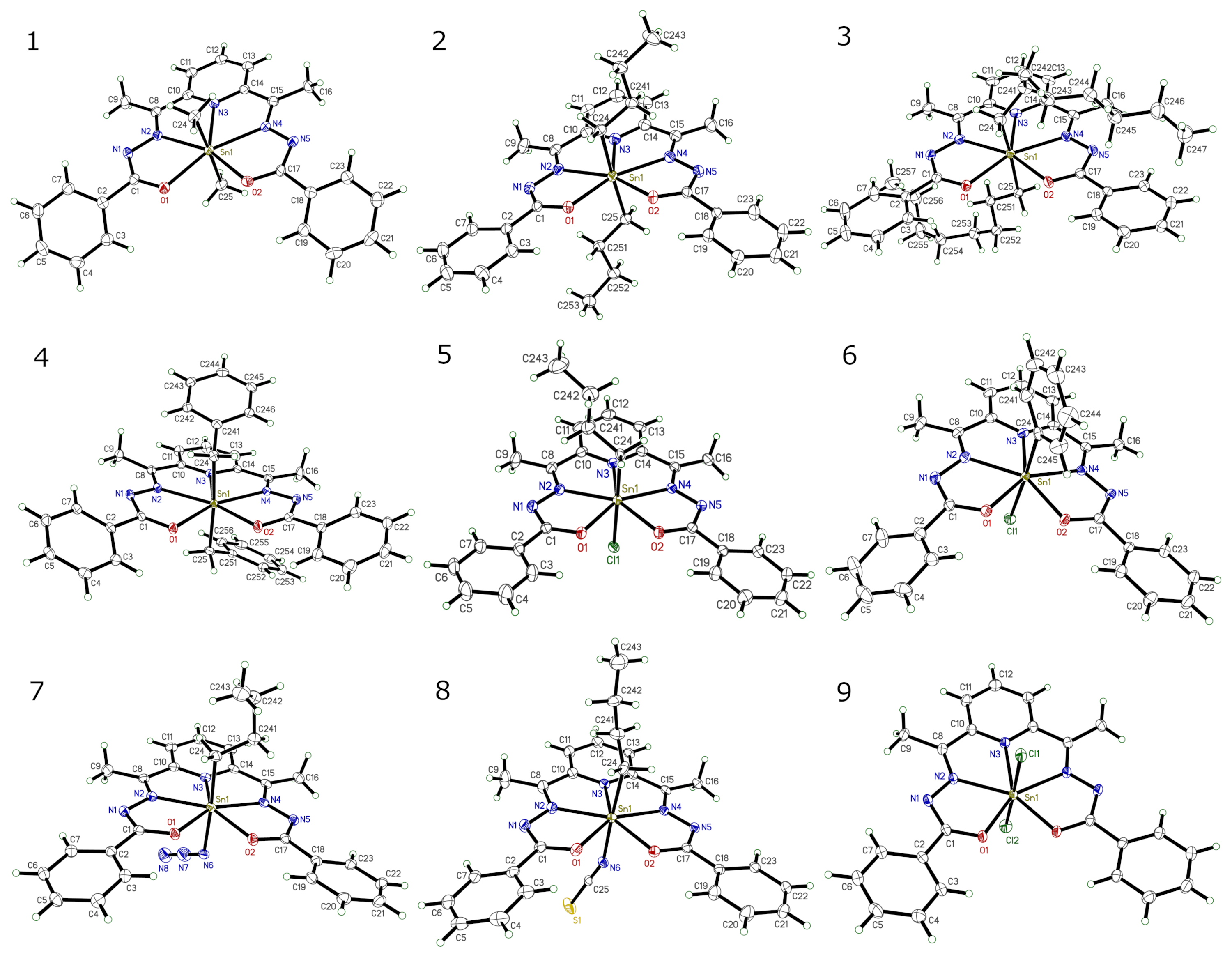
As with the previous example, these all have the same ligand attached to their central Sn atom, with two other groups attached. These are: two methyl (1), two butyl (2), two octyl (3), two benzyl (4), butyl and chloro (5), phenyl and chloro (6), butyl and azido (7), butyl and thiocyanato (8), and two chloro (9). For clarity, we'll pare these groups down to the first atom. One additional complication is that the structure of (9) straddles the mirror plane of Pnma, so we'll have to generate the full model first.
The CIFs and other files for this example are available here.
For structures 1-8, over-rip.py does a good job. Structures 2, 5, 8 have disorder so we'll take the major component only. Solvent molecules present in 5 and 9 will also need to be manually edited out. For structure 9, 'GROW' in Shelxtl XP will generate the whole molecule, which may then be written to a file using 'ORTH' or 'FILE'd and converted to CIF using Mercury. The ORTH-generated file is easily edited to .xyz format. Other methods would also work. Over-rip.py is run in the usual way, viz:
./over-rip.py or python over-rip.py
The result is nine .xyz files, copies of which are here.
The nine Sn complexes each have 33 atoms (after editing the ligands). Since there could be torsion of the phenyl rings, only their ipso carbons make sense to include in the fit. Similarly, since not all of the pendant groups bond to Sn via carbon, it would be a mistake to include them in the fit. Thus, we'll feed over-rot.py an atom list using an over-rot-atoms.txt file with the following atoms:
Sn1 O1 O2 N1 N2 N3 N4 N5 C1 C2 C8 C9 C10 C11 C12 C13 C14 C15 C16 C17 C18
With the nine .xyz files in the same directory as over-rot.py, a successful run using:
./over-rot.py or python over-rot.py
Leads to the following output, which is written to a log file 'over-rot.log'.
Using reference structure: 1.xyz
Using the following atoms for fitting: Sn1, O1, O2, N1, N2, N3, N4, N5, C1, C2, C8, C9, C10, C11, C12, C13, C14, C15, C16, C17, C18
All specified atoms found in reference structure
=== Pre-alignment Analysis ===
Targeted Atoms RMSD Matrix (Å):
-----------------------------------------------------------------------------------
1 2 3 4 5 6 7 8 9
-----------------------------------------------------------------------------------
1 1.xyz 0.000 8.553 6.361 7.726 12.708 12.701 10.485 12.654 10.578
2 2.xyz 8.553 0.000 6.417 12.636 9.511 9.197 7.434 9.340 7.950
3 3.xyz 6.361 6.417 0.000 10.101 11.222 11.620 8.256 9.727 10.125
4 4.xyz 7.726 12.636 10.101 0.000 18.638 18.340 13.969 17.633 15.778
5 5.xyz 12.708 9.511 11.222 18.638 0.000 4.415 9.506 5.772 7.262
6 6.xyz 12.701 9.197 11.620 18.340 4.415 0.000 9.139 6.086 6.318
7 7.xyz 10.485 7.434 8.256 13.969 9.506 9.139 0.000 7.293 9.838
8 8.xyz 12.654 9.340 9.727 17.633 5.772 6.086 7.293 0.000 10.023
9 9.xyz 10.578 7.950 10.125 15.778 7.262 6.318 9.838 10.023 0.000
-----------------------------------------------------------------------------------
Global RMSD (pre-fitted atoms): 4.1243 Å
=== Alignment ===
Note: Inverted coordinates give better RMSD (0.1144 Å vs 0.1760 Å)
Use inverted coordinates for this structure? (y/n): Aligned 2.xyz to reference | Fit RMSD: 0.1144 Å | Rotation: 93.97° (inverted)
Note: Inverted coordinates give better RMSD (0.0969 Å vs 0.1081 Å)
Use inverted coordinates for this structure? (y/n): Aligned 3.xyz to reference | Fit RMSD: 0.0969 Å | Rotation: 120.18° (inverted)
Aligned 4.xyz to reference | Fit RMSD: 0.1642 Å | Rotation: 142.31°
Note: Inverted coordinates give better RMSD (0.1437 Å vs 0.2101 Å)
Use inverted coordinates for this structure? (y/n): Aligned 5.xyz to reference | Fit RMSD: 0.1437 Å | Rotation: 171.53° (inverted)
Note: Inverted coordinates give better RMSD (0.2199 Å vs 0.2567 Å)
Use inverted coordinates for this structure? (y/n): Aligned 6.xyz to reference | Fit RMSD: 0.2199 Å | Rotation: 99.51° (inverted)
Note: Inverted coordinates give better RMSD (0.1710 Å vs 0.1992 Å)
Use inverted coordinates for this structure? (y/n): Aligned 7.xyz to reference | Fit RMSD: 0.1710 Å | Rotation: 100.99° (inverted)
Note: Inverted coordinates give better RMSD (0.1734 Å vs 0.2092 Å)
Use inverted coordinates for this structure? (y/n): Aligned 8.xyz to reference | Fit RMSD: 0.1734 Å | Rotation: 110.20° (inverted)
Aligned 9.xyz to reference | Fit RMSD: 0.2096 Å | Rotation: 167.68°
Overall Rotation Angles:
----------------------------------------------------
Index Filename Angle (°) Inverted
----------------------------------------------------
1 1.xyz 0.00 No
2 2.xyz 93.97 Yes
3 3.xyz 120.18 Yes
4 4.xyz 142.31 No
5 5.xyz 171.53 Yes
6 6.xyz 99.51 Yes
7 7.xyz 100.99 Yes
8 8.xyz 110.20 Yes
9 9.xyz 167.68 No
----------------------------------------------------
All aligned structures written to over-rot.rot
=== Post-alignment Analysis ===
Rotated Atoms RMSD Matrix (Å):
-----------------------------------------------------------------------------------
1 2 3 4 5 6 7 8 9
-----------------------------------------------------------------------------------
1 1.xyz 0.000 0.114 0.097 0.164 0.144 0.220 0.171 0.173 0.210
2 2.xyz 0.114 0.000 0.135 0.095 0.108 0.181 0.117 0.141 0.154
3 3.xyz 0.097 0.135 0.000 0.198 0.165 0.229 0.180 0.179 0.219
4 4.xyz 0.164 0.095 0.198 0.000 0.154 0.208 0.140 0.182 0.185
5 5.xyz 0.144 0.108 0.165 0.154 0.000 0.127 0.068 0.084 0.092
6 6.xyz 0.220 0.181 0.229 0.208 0.127 0.000 0.105 0.079 0.070
7 7.xyz 0.171 0.117 0.180 0.140 0.068 0.105 0.000 0.060 0.081
8 8.xyz 0.173 0.141 0.179 0.182 0.084 0.079 0.060 0.000 0.082
9 9.xyz 0.210 0.154 0.219 0.185 0.092 0.070 0.081 0.082 0.000
-----------------------------------------------------------------------------------
Global RMSD (rotated atoms): 0.0578 Å
Improvement: 4.0666 Å
In this example, six of the nine were inverted. The space-group symmetry for 6 was P 212121, so one might expect inversion to be invalid. In actuality, the crystals of 6 were twinned by inversion, so the inverted form was present. Either way, the molecule of 6 is achiral and (chemically, not crystallographically) mirror symmetric, so it makes no difference. In fact, if 6 had been numbered the other way round it wouldn't have suggested inversion (likewise for 2, 3, 5, 7, 8).
Ok, on to the 3D graphics. If we feed the new .rot file to over-lay.py :
./over-rot.py or python over-rot.py
The resulting html/JavaScript should look like this:
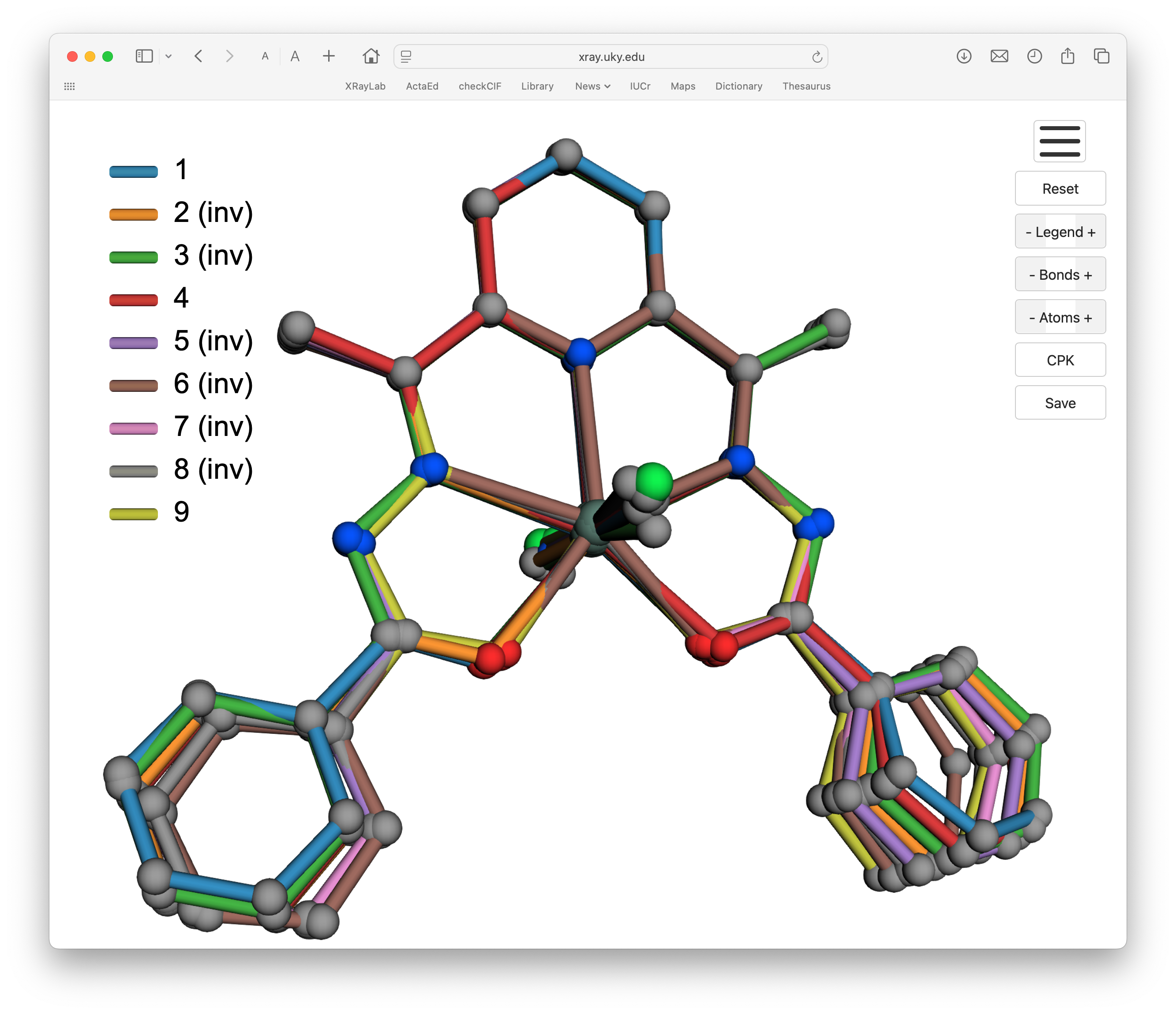
This example introduced yet more complexity. One of the structures had to be grown to get its full complement of atoms due to it being on a crystallographic mirror plane. Nevertheless, given proper attention to detail, the procedure works well.
2: Extract atom coordinates from the CIF(s)
3: Optimal superposition via quaternions
4: 3D interactive graphics
5: Three tin complexes and their pro-ligand
6: Nine tin complexes
7: Seventeen arsenic complexes