5/7: Three tin complexes and their pro-ligand
In the following example we'll superimpose three Sn(IV) complexes and their pro-ligand from a recent paper. As with the metconazole example, an overlay plot made using Mercury was included in the paper.
Let's first take a look at the four structures:
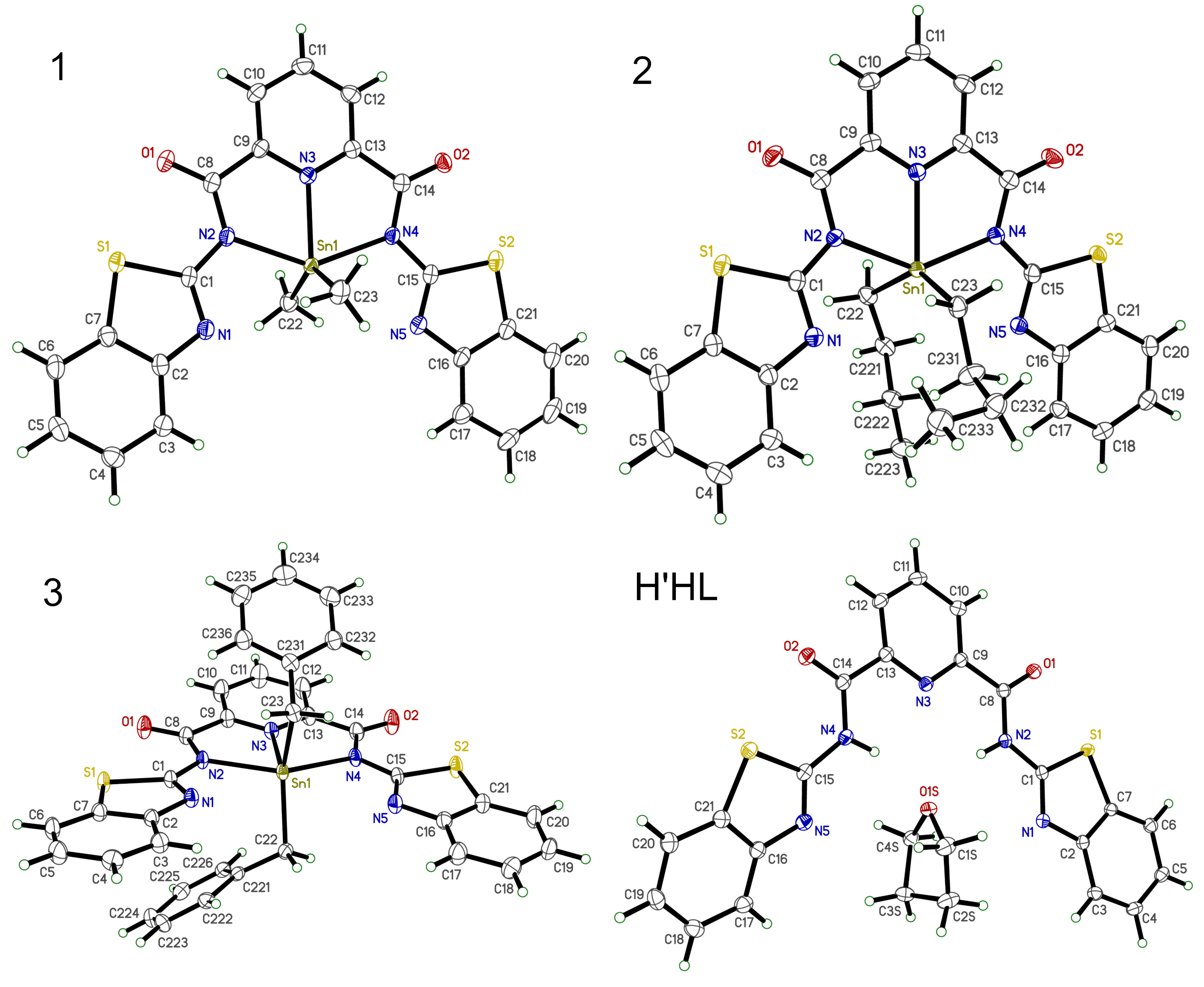
Structures 1, 2, and 3 have the same ligand bound to the tin atom, but have other additional substituents attached: two methyls in 1, two butyls in 2, and two benzyls in 3. For the sake of clarity, these groups will be trimmed to just their first carbon. The ligand structure H'HL has a THF solvent H-bonded to it, but we'll cut that out as well. Note that the numbering schemes were carefully chosen so that equivalent atoms in each structure have the same atom name. It does not matter if editing of the structure files is done on the CIFs, the .xyz, or .rot files.
The CIFs and other files for this example are available here.
The first step is to generate .xyz files for each of the CIFs. On a Mac or Linux, if the CIFs are in the same directory as over-rip.py, then run the program in the usual way. From a terminal, either of the following:
./over-rip.py
python over-rip.py
will work ... not sure about Windows. The result should be four .xyz files, copies of which are here. Note that structure 2 had disorder of one butyl. Taking just the major component in 2, our interaction with over-rip.py looks like this:
Found 4 CIF file(s): - 1.cif - 2.cif - 3.cif - H'HL.cif Processing 1.cif... Done (33 atoms, all occupancies=1) Processing 2.cif... Found atoms with fractional occupancy. Keep minor components (<0.5) and full occupancy (1.0) atoms? [y/N]: Done (33 atoms, major) Processing 3.cif... Done (33 atoms, all occupancies=1) Processing H'HL.cif... Done (30 atoms, all occupancies=1) All files processed.
The three Sn complexes each have 33 atoms (after truncating the alkyl and benzyl groups), while the pro-ligand has 30. Since there could be torsion about the C1-N2 and C15-N4 bonds, it is best to not include the whole of each benzothiazole group. Thus, for over-rot.py it would be bad to use 'all' to specify which atoms to fit. Likewise, it would be a mistake to use just the first n atoms unless we rearranged the atom sequences. One solution is to supply a list of atoms to fit, either in response to being asked by over-rot.py or by making a file 'over-rot-atoms.txt' with a bunch of atom labels, like this:
N1 N2 N3 N4 N5 O1 O2 C1 C2 C8 C9 C10 C11 C12 C13 C14 C15 C16 C17 C18
With these atoms, we'll fit the central part of the ligand, but only C1, N1 and C15, N5 of the benzothiazole groups. Due to the torsions mentioned above, we could justifiably also skip N1 and N5 (try it and see). Anyhow, with the four .xyz files in the same directory as over-rot.py, run the program in the usual way, one of these:
./over-rot.py
python over-rot.py
This leads to the following output by over-rot.py, which is also written to a log file 'over-rot.log'.
Using reference structure: 1.xyz
Using the following atoms for fitting: N1, N2, N3, N4, N5, O1, O2, C1, C2, C8, C9, C10, C11, C12, C13, C14, C15, C16, C17, C18
All specified atoms found in reference structure
Using 20 atoms for fitting
=== Pre-alignment Analysis ===
Targeted Atoms RMSD Matrix (Å):
--------------------------------------------------------
1 2 3 4
--------------------------------------------------------
1 1.xyz 0.000 12.097 13.059 8.712
2 2.xyz 12.097 0.000 18.680 12.763
3 3.xyz 13.059 18.680 0.000 11.154
4 H'HL.xyz 8.712 12.763 11.154 0.000
Global RMSD (pre-fitted atoms): 4.6302 Å
=== Alignment ===
Aligned 2.xyz to reference | Fit RMSD: 0.1678 Å | Rotation: 130.66°
Aligned 3.xyz to reference | Fit RMSD: 0.0864 Å | Rotation: 178.95°
Note: Inverted coordinates give better RMSD (0.4791 Å vs 0.5549 Å)
Use inverted coordinates for this structure? (y/n): Aligned H'HL.xyz to reference | Fit RMSD: 0.4791 Å | Rotation: 62.48° (inverted)
Overall Rotation Angles:
----------------------------------------------------
Index Filename Angle (°) Inverted
----------------------------------------------------
1 1.xyz 0.00 No
2 2.xyz 130.66 No
3 3.xyz 178.95 No
4 H'HL.xyz 62.48 Yes
All aligned structures written to over-rot.rot
=== Post-alignment Analysis ===
Rotated Atoms RMSD Matrix (Å):
--------------------------------------------------------
1 2 3 4
--------------------------------------------------------
1 1.xyz 0.000 0.168 0.086 0.479
2 2.xyz 0.168 0.000 0.174 0.347
3 3.xyz 0.086 0.174 0.000 0.501
4 H'HL.xyz 0.479 0.347 0.501 0.000
Global RMSD (rotated atoms): 0.1178 Å
Improvement: 4.5123 Å
A surprise here was seeing that the H'HL structure gave a better fit when inverted! It's not by much because the molecule is fairly flat, but this is a check that is not automated in Mercury.
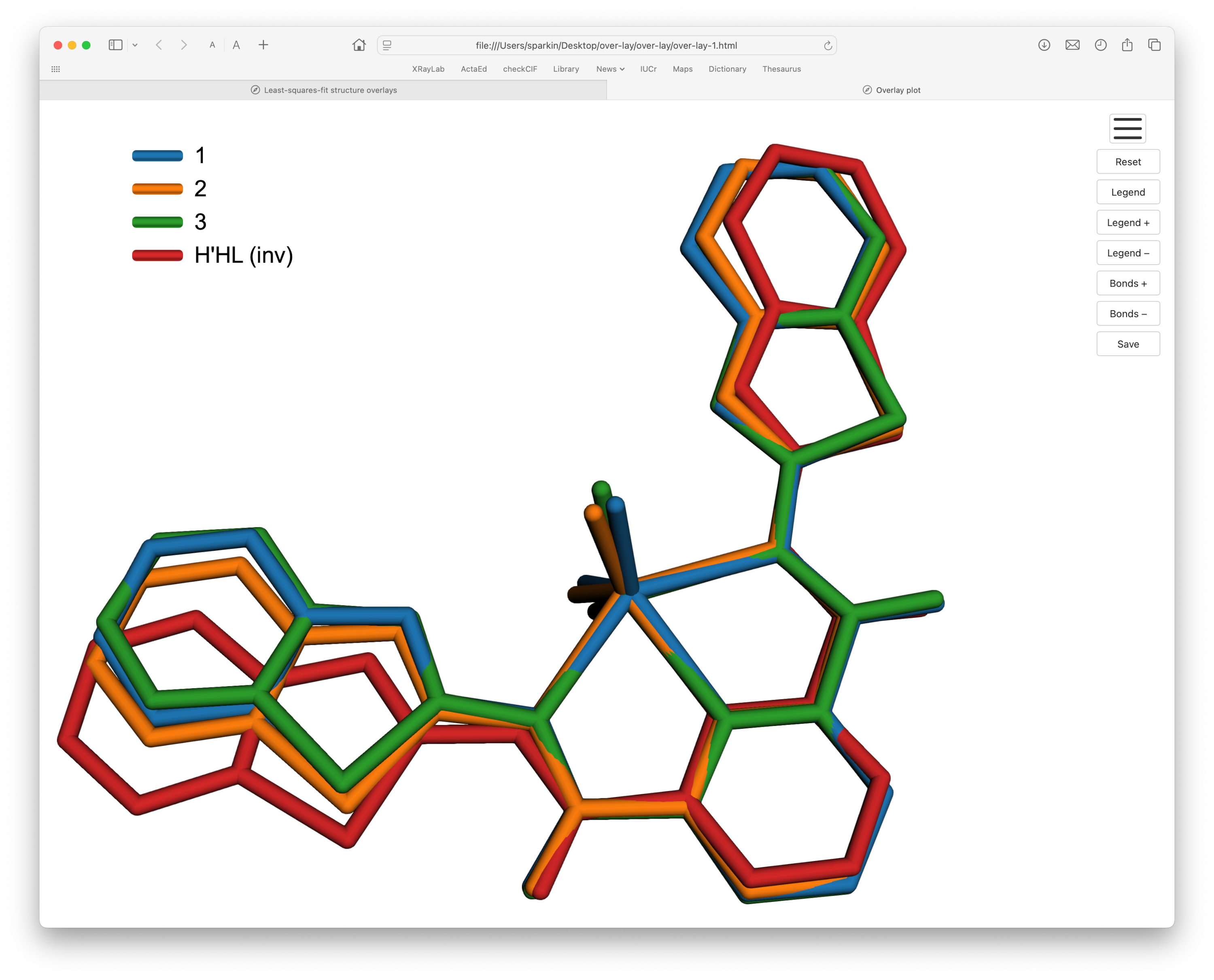
The next step is, of course, the eye candy. With the newly generated .rot file (re-named to 3Sn-plus-ligand.rot, or whatever) moved to where over-lay.py can find it, we get the html/JavaScript file, ready to be viewed in a browser, something like this:
If for some for some reason you wanted an overlay with the THF solvent solvent molecule that is hydrogen bonded to the H'HL ligand, that too is possible:
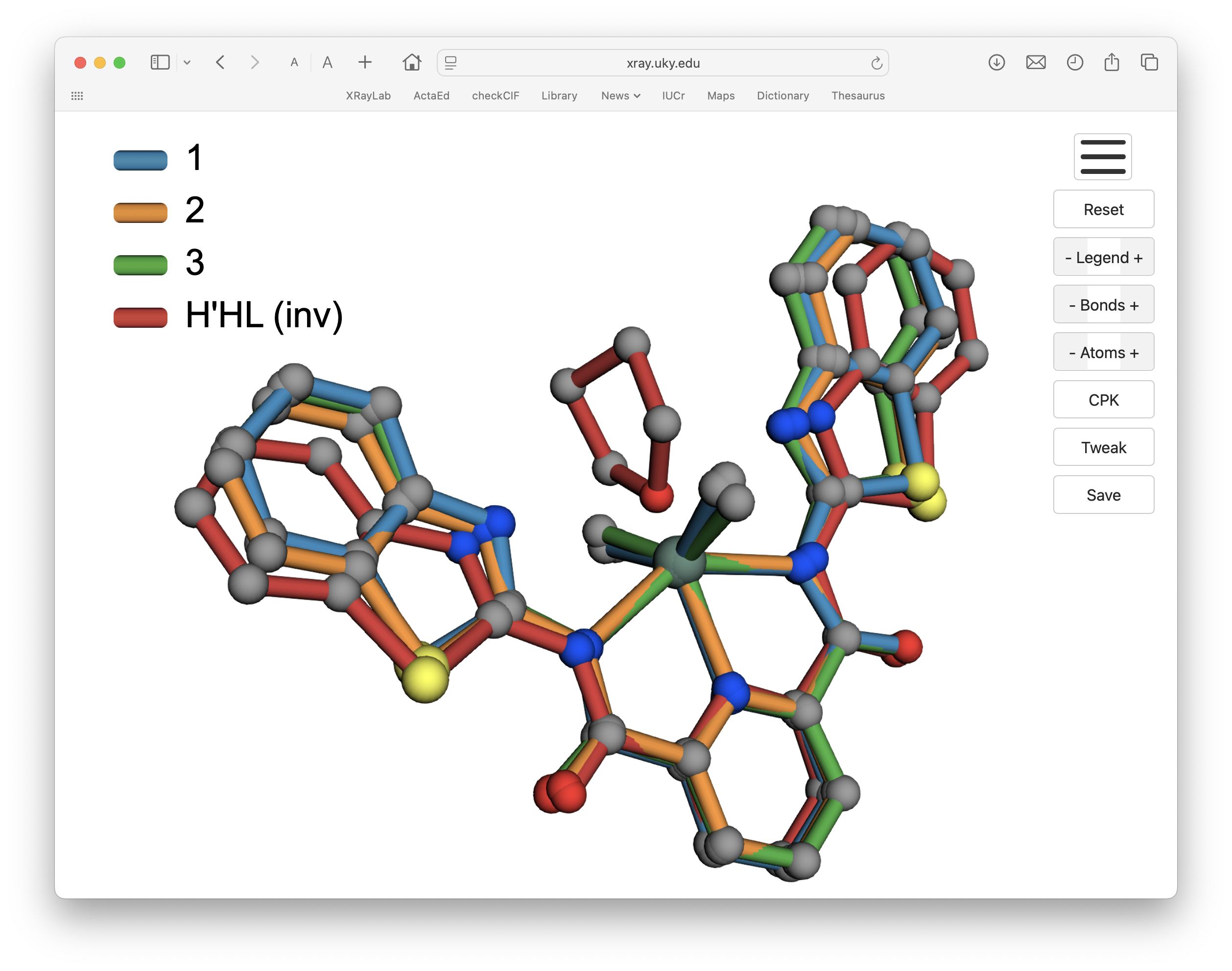
This example introduced a bit more complexity than the metconazole overlay in sections 1-4. One structure had a different atom count from the others and also fit marginally better when inverted. The end result, however, works in much the same way.
2: Extract atom coordinates from the CIF(s)
3: Optimal superposition via quaternions
4: 3D interactive graphics
5: Three tin complexes and their pro-ligand
6: Nine tin complexes
7: Seventeen arsenic complexes