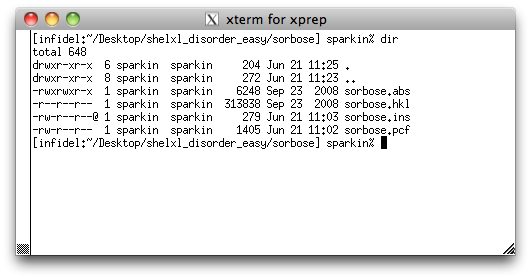
Notice that there are two other files: sorbose.abs and sorbose.pcf.
The '.abs' file has details of the SADABS run (data merging and absorption
correction) and the '.pcf' has a few experimental details in CIF format. They were
created as part of the data reduction process, but we will not use them in this
tutorial.
Let's take a look inside the '.ins' file to re-familiarize ourselves with the instructions needed to solve a structure with SHELXS. You can use any text editor you like, but as with the sucrose tutorial, we will use the nano editor, so type 'nano sorbose.ins' at the prompt, and hit <return>. You should see something like this:
Let's take a look inside the '.ins' file to re-familiarize ourselves with the instructions needed to solve a structure with SHELXS. You can use any text editor you like, but as with the sucrose tutorial, we will use the nano editor, so type 'nano sorbose.ins' at the prompt, and hit <return>. You should see something like this:
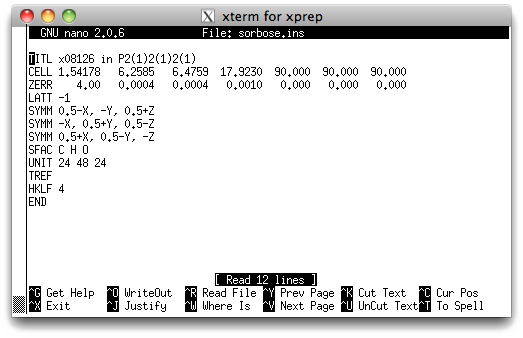
You should have seen lines like these already in the sucrose tutorial. Briefly,
TITL - Change 'x08126' to 'sorbose' etc. if you like.
CELL - X-ray wavelength followed by the unit cell parameters.
ZERR - Z, followed by uncertainties on the cell parameters.
LATT - Bravais lattice. A value of -1 means primitive non-centrosymmetric.
SYMM - Symmetry equivalent positions. Here it's three 21screw axes.
SFAC - Indicates what atoms are present.
UNIT - Number of each atom type in the unit cell.
TREF - Tangent REFinement, this instruction solves the structure.
HKLF - Reads the x-ray data, '4' specifies the format of the data.
END - marks the end of the instructions used by SHELXS.
CELL - X-ray wavelength followed by the unit cell parameters.
ZERR - Z, followed by uncertainties on the cell parameters.
LATT - Bravais lattice. A value of -1 means primitive non-centrosymmetric.
SYMM - Symmetry equivalent positions. Here it's three 21screw axes.
SFAC - Indicates what atoms are present.
UNIT - Number of each atom type in the unit cell.
TREF - Tangent REFinement, this instruction solves the structure.
HKLF - Reads the x-ray data, '4' specifies the format of the data.
END - marks the end of the instructions used by SHELXS.
To solve the structure, simply type 'shelxs sorbose' at the prompt and hit
<return>, like this:

Structures with minor disorder are usually no more difficult to solve than
non-disordered structures. To a trained eye it is clear from the above output that
the structure is solved. Successful solutions are usually indicated by small CFOM,
NQUAL values close to -1, and RE values less than about 0.25. If you now do a directory
listing you will see two new files: sorbose.lst and sorbose.res. The
'.lst' file is a listing of what happened during the SHELXS run. It contains
a wealth of useful information, but much of it is too specialized to worry about yet.
The file we are interested in is the '.res' file, which if you remember contains
results ('.ins' = instructions, '.res' = results).
You can look at the '.res' file with a text editor (e.g. nano) if you like, but it probably won't mean much yet. If you choose to look, it should be very similar to this:
You can look at the '.res' file with a text editor (e.g. nano) if you like, but it probably won't mean much yet. If you choose to look, it should be very similar to this:
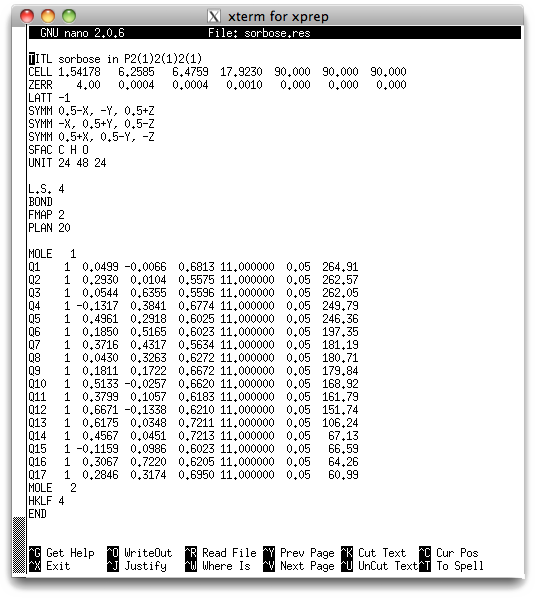
The meaning of the file contents is given in the sucrose tutorial, so it will not be
repeated here. One important thing to notice is that SHELXS found 17 peaks in
the E-map that it tentatively thinks could be potential atom sites. Five of
them (Q1-Q5) have peak heights of about 240 or above, seven are between about 150
and 200, one is about 100, and four are in the 60s. In an E-map, the
relative peak heights can be useful, but the absolute numbers are not.
The five largest peaks are likely to be oxygen, and the smaller peaks are quite likely
to be noise. However, since we know there is disorder in this structure we will have
to pay close attention to the actual peaks and their positions. Dealing with disorder
means that you have to identify partially occupied atom sites. Such partial
atoms obviously have less electron density, and hence smaller than expected peak
heights than ordinary atoms. We will use XP from SHELXTL to edit the
initial structure solution.
Return to the first page of this tutorial
Return to the main Tutorials page or to the main X-Ray Lab page
Return to the main Tutorials page or to the main X-Ray Lab page