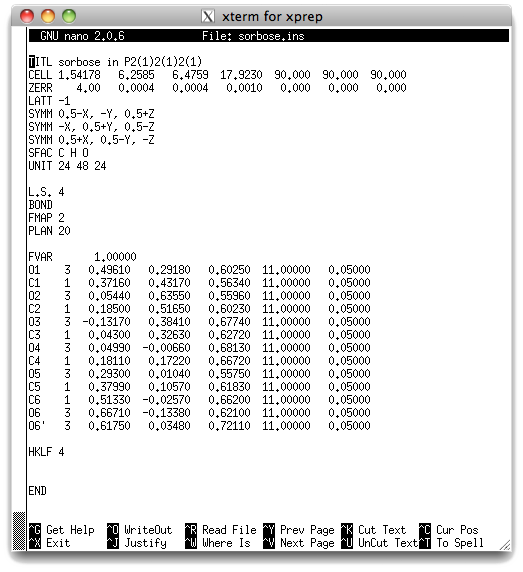
This file will perform four cycles of least-squares refinement on the model. Most of
the structure should refine quite well, but our disordered atoms (O6 & O6') should
cause a problem because they still have full occupancy. The occupancy factor is given
in column 6 of the atom list - notice that they are all 11.00000, which is the
SHELXL way of stating "the occupancy of this atom is exactly 1.00000, and
it should not be refined (adding 10 to a parameter fixes its value). What do you
think will happen to O6 and O6' if we refine them like this ? Let's find out: exit
from the editor and type shelxl sorbose <return>.

Notice that the wR2 value drops, but it seems to hang at a quite high value (38.25%),
and that R1 is also pretty high (11.45%). It is quite likely that the refinement has
not quite converged, but at this stage that is not much of a problem. Let's look in
the '.res' file and see if there is anything unusual about the atoms we think are
disordered (O6 & O6'):

Notice that the displacement parameters (last column) of O6 and O6' are roughly twice
and three times as big as those of the rest of the atoms. Clearly something is not
right. The effect can be even more dramatic if we look at an ellipsoid plot. You
could use XP for this, but let's try something different -
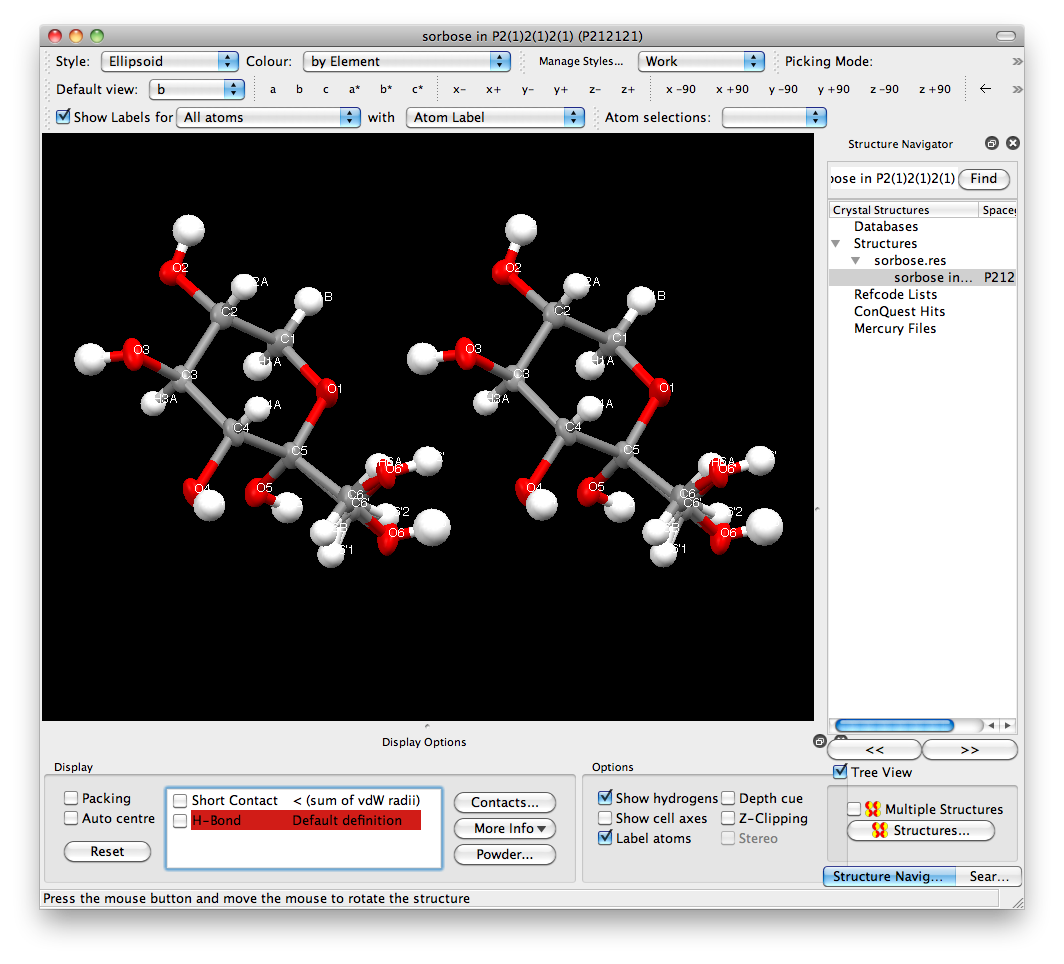
Mercury, which is an
excellent free program from the CCDC (it is
a good idea to familiarize yourself with a wide variety of programs). The Mercury
view looks like this:
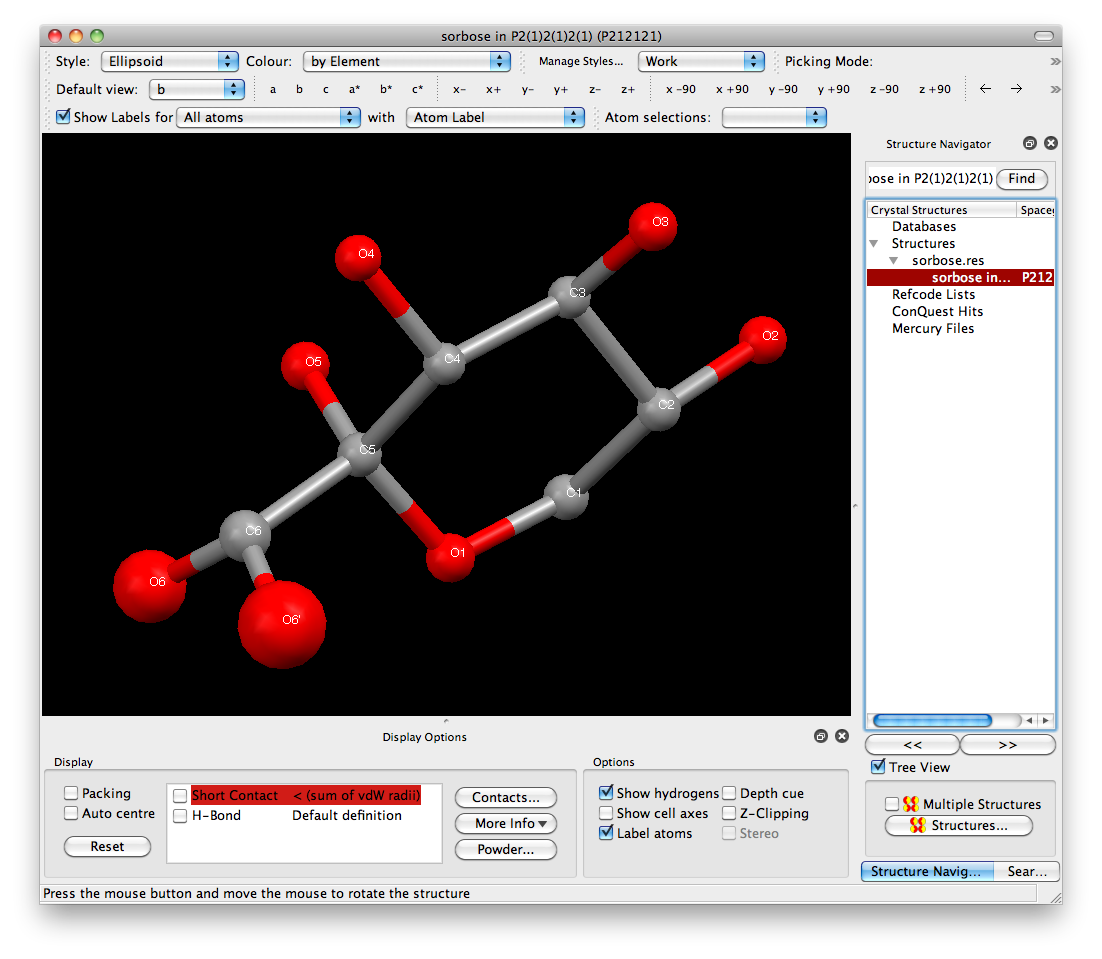
The balls for O6 and O6' are clearly larger than the rest of the atoms. The reason
is quite straightforward if you think about it. Since O6 and O6' have partial
occupancy, neither of them has the scattering power of a full oxygen atom. Remember,
the refinement program alters the model to try to make it fit the experimental data
better. In this case, the only thing that SHELXL is able to do to make the
disordered atoms fit better is to smear them out to make the full occupancy oxygens
in the model fit the fractional occupancy oxygens that were in the crystal. It does
this by increasing their displacement parameters, and that is why the numbers in
column 7 for O6 and O6' are much higher than for the rest of the atoms. It should
also follow from this that O6 is the major and O6' the minor component. With more
complicated disorder, it is not unusual for initial assigments of major and minor to
be incorrect, but that is not the case here. Let's go ahead and fix up the '.res'
file to improve the disorder model.

A few changes have been made. Some are just general commands that you would add to
any structure:
TEMP - temperature in celsius.
SIZE - crystal size in mm.
L.S. - increased to 6 (disordered structures take more cycles to converge.
BOND $H - includes H atoms in bond length etc. tables
CONF - include torsion angles in the CIF
HTAB - write hydrogen bond information to the '.lst' file
ACTA - write a CIF.
SIZE - crystal size in mm.
L.S. - increased to 6 (disordered structures take more cycles to converge.
BOND $H - includes H atoms in bond length etc. tables
CONF - include torsion angles in the CIF
HTAB - write hydrogen bond information to the '.lst' file
ACTA - write a CIF.
The rest of the changes describe the disorder model. Disordered groups have been
split into two PARTs. PART 1 contains C6 and O6, while PART 2 has a copy of
C6 (renamed to C6') and O6'. The PART 0 command indicates the end of this disordered
region (in a more complicated structure, there could be several disordered regions).
The occupancy factor of the PART 1 atoms has been changed to 21.00000, while that of
PART 2 atoms has been changed to -21.00000. This may seem unusual, but it is quite
logical, and it's the standard way of dealing with occupancies of two-part disorder
in SHELXL. The '2' in '21.00000' refers to the second number on the so-called
free-variable or FVAR instruction, which has been set so 0.6 (you could
choose a different fraction, as it will refine, but it helps to make a reasonably
good guess). The way these occupancies are interpreted in SHELXL is that
'21.00000' means 'second FVAR number, multiplied by one', and '-21.00000'
means 'one minus the second FVAR number, all multiplied by one'. In this way,
the PART 1 atoms get occupancy 0.6, while the PART 2 atoms get occupancy 0.4. Notice
that these two values add up to exactly one, which ensures that for all disorder
components combined, we get the right atom count. In a more complicated structure
with several disordered groups, it may be necessary to use the 3rd, 4th, 5th and
so on FVAR numbers to define different occupancies. Also, some disorder (like
partial occupancy disordered solvent) is non-stoichiometric.
The other additions to the file are constraints (EXYZ and EADP) and restraints (SAME and SIMU). In the crystallographic sense, a constraint is an exact mathematical relationship that allows two or more quantities to use the same variable, while a restraint is a means of adding extra information to the refinement. In the present structure, we have split atom C6 into two PARTs (C6 and C6'), but both parts are superimposed. At least for now, we need to ensure that the coordinates and the displacement parameters of C6 and C6' keep the same values. For this we use EXYZ (Equal XYZ coordinates) and EADP (Equal Anisotropic Displacement Parameters). You may be wondering why we bothered to split C6 at all! The answer to that lies in the placement of methylene hydrogen atoms between the two disordered CH2OH groups. Since the oxygen atom is disordered over two positions, the H atoms on the methylene must also be disordered. Splitting C6 makes it much easier to add these disordered H atoms in a sensible way. Later, we will attempt to relax the EXYZ criterion, as sometimes this is justifiable. The EADP C6 C6' constraint, however, is non-negotiable: this constraint is always required for overlapping partial atoms. The SAME restraint ensures that the geometry (bond lengths and angles) are the same for the sequence of atoms following the command (i.e. C6', O6') as they are for the sequence of atoms on the lines below the command (i.e. C6, O6). We know that this is reasonable because both major and minor components are chemically identical. The SIMU command restrains the displacement parameters of the named atoms (i.e. all those from C6 to O6' in the atom list) to have similar values. Again, this is reasonable given the proximity of the atoms, but we will change this restraint later when we include anisotropic temperature factors.
When we refine this model (i.e. save it as an '.ins' file, then type shelxl sorbose <return> at the xterm prompt), this is what we get:
The other additions to the file are constraints (EXYZ and EADP) and restraints (SAME and SIMU). In the crystallographic sense, a constraint is an exact mathematical relationship that allows two or more quantities to use the same variable, while a restraint is a means of adding extra information to the refinement. In the present structure, we have split atom C6 into two PARTs (C6 and C6'), but both parts are superimposed. At least for now, we need to ensure that the coordinates and the displacement parameters of C6 and C6' keep the same values. For this we use EXYZ (Equal XYZ coordinates) and EADP (Equal Anisotropic Displacement Parameters). You may be wondering why we bothered to split C6 at all! The answer to that lies in the placement of methylene hydrogen atoms between the two disordered CH2OH groups. Since the oxygen atom is disordered over two positions, the H atoms on the methylene must also be disordered. Splitting C6 makes it much easier to add these disordered H atoms in a sensible way. Later, we will attempt to relax the EXYZ criterion, as sometimes this is justifiable. The EADP C6 C6' constraint, however, is non-negotiable: this constraint is always required for overlapping partial atoms. The SAME restraint ensures that the geometry (bond lengths and angles) are the same for the sequence of atoms following the command (i.e. C6', O6') as they are for the sequence of atoms on the lines below the command (i.e. C6, O6). We know that this is reasonable because both major and minor components are chemically identical. The SIMU command restrains the displacement parameters of the named atoms (i.e. all those from C6 to O6' in the atom list) to have similar values. Again, this is reasonable given the proximity of the atoms, but we will change this restraint later when we include anisotropic temperature factors.
When we refine this model (i.e. save it as an '.ins' file, then type shelxl sorbose <return> at the xterm prompt), this is what we get:

The R-values have come down a bit and the difference map is a little flatter, but
nothing too dramatic seems to have happened. You could look at the model with XP
or Mercury if you like. You could also look at the '.res' file in a text editor,
which will show you that the displacement parameters for the disordered atoms are now
much more similar to the ordered atoms. At this point we could make the model
anisotropic by adding a command ANIS and then running SHELXL again. If
you do this, the SHELXL screen output should look similar to this:

The improvement is a whole lot more dramatic! However, the SIMU restraint is
not entirely appropriate in this case because with anisotropic displacement parameters
(ADPs) it tends to make all the restrained ellipsoids point in the same direction. A
better alternative would be DELU, which restrains the components of ADPs along
bonds to be similar (DELU is sometimes called a rigid-bond restraint, or
a Hirshfeld restraint). Let's go ahead and look for H atoms in the difference
map, and for the next round we'll remember to change SIMU to DELU. In
XP, the structure and the top 11 difference map peaks look like this:
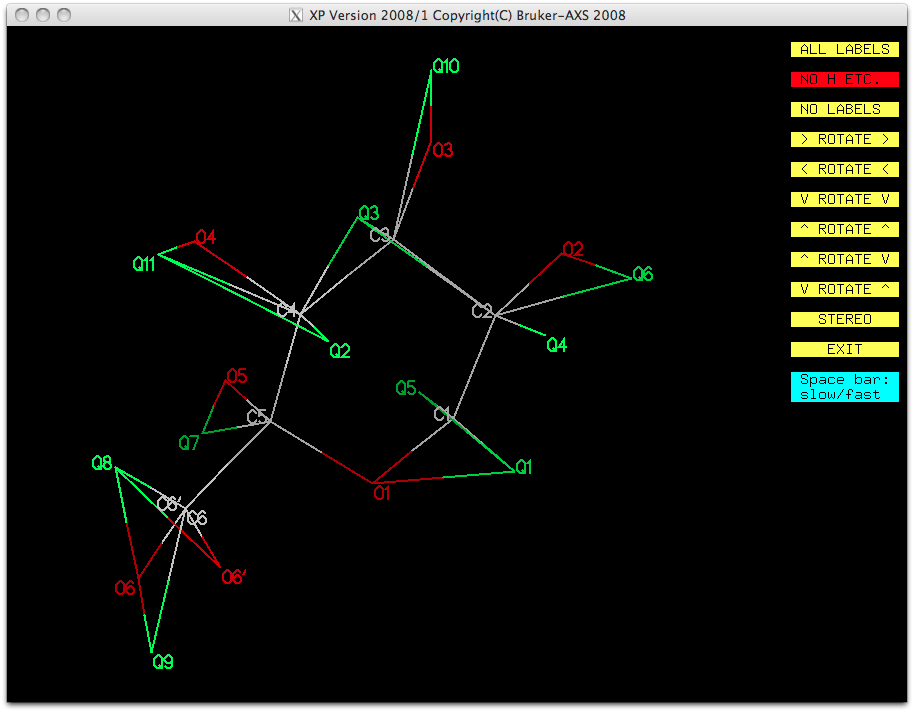
It looks as though all the CH and CH2 groups except for C6 & C6' are
present, as are all of the OH hydrogens except O6 & O6'. Add these to the model
with the following commands:
HFIX 13 c2 c3 c4
HFIX 23 c1
HFIX 147 o2 o3 o4 o5
HFIX 23 c1
HFIX 147 o2 o3 o4 o5
Refinement with SHELXL gives the following screen output:

That's another dramatic improvement. We are now in a position to fine-tune the
disorder model and make other small changes like weighting scheme tweaks, test for
extinction, check the absolute configuration via the Flack parameter etc.
To see if we can add hydrogens to the disordered groups, load the '.res' file into
XP. The difference map should be substantially flat (check the '.res' file or
use the info command in XP). In the following image, difference map
peaks Q6 to Q20 have been eliminated.
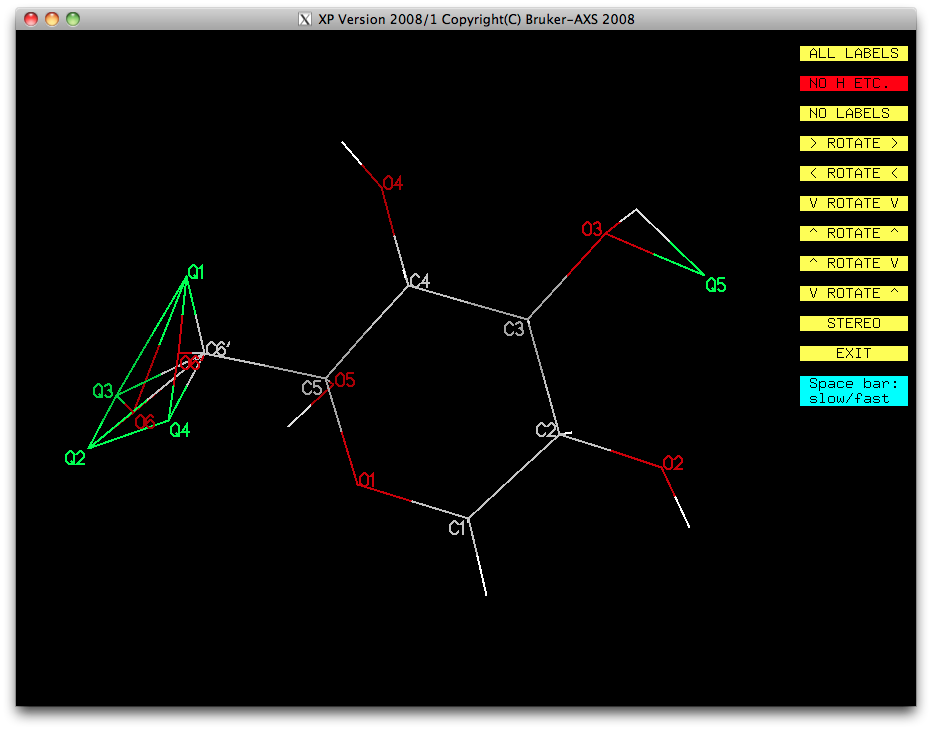
In the above, Q5 is clearly garbage so we will ignore it. It looks as though there is
some residual electron density around the disordered atoms that could be attributed to
hydrogen. In this example, we can model the split CH2 groups easily using
the HFIX 23 instruction. The only difficulty are the disordered hydroxyl H atoms.
Notice that we are making only small changes to the model and then refining. This is
generally the best strategy when trying to eke out fine features, especially in a
disordered structure. For the next round of refinement we will further edit the '.res'
and make a '.ins' file with the following additions/changes:
HFIX 23 C6 C6'
Update the weighting scheme
Test for extinction
Attempt to free the EXYZ constraint on C6 & C6' (type 'REM' in front of the command)
Test for extinction
Attempt to free the EXYZ constraint on C6 & C6' (type 'REM' in front of the command)
Addition of partial hydrogens, especially to hydroxyl groups, is often of questionable
utility, but in this structure it does seem reasonable to assign the biggest difference
map peak as the H atom on O6. The second biggest also looks like it could be the H on
O6', but it is very small, only 0.16 eÅ-3. It is usually not a good
idea to put too much credence in such tiny details, but if they make chemical sense
you may include them: just be prepared to defend your model! So, we should be able to
add a partial H to O6 with an HFIX 147 command. O6' is more of a problem, but HFIX 87
may be possible because atom O4 of a symmetry-related molecule is positioned to make
a reasonable hydrogen bond to both O6 and O6', as you can see from this Mercury
image:
The eagle-eyed amongst you may notice that the model needs to be inverted (this is
naturally occurring sorbose). You can achieve this with the command 'MOVE 1 1 1 -1'
(see the manual for details). If you add both of these partial hydrogens in the way
described, and set the correct chirality, you should end up with the following final
model:
That's the end of this tutorial. It went into a little finer detail than originally
intended, but it is all information you need to know. The main point to remember is
that disorder has to make good chemical sense. If your disorder model does not make
chemical sense then it is very unlikely to be correct.
Return to the first page of this tutorial
Return to the main Tutorials page or to the main X-Ray Lab page
Return to the main Tutorials page or to the main X-Ray Lab page