A run of SHELXS in the normal way gives the following screen output:

Note that Ralpha, Nqual and the CFOM all look pretty good. These
statistics, along with the sub 25% RE value indicate a likely solution. In XP
we get the following E-map:
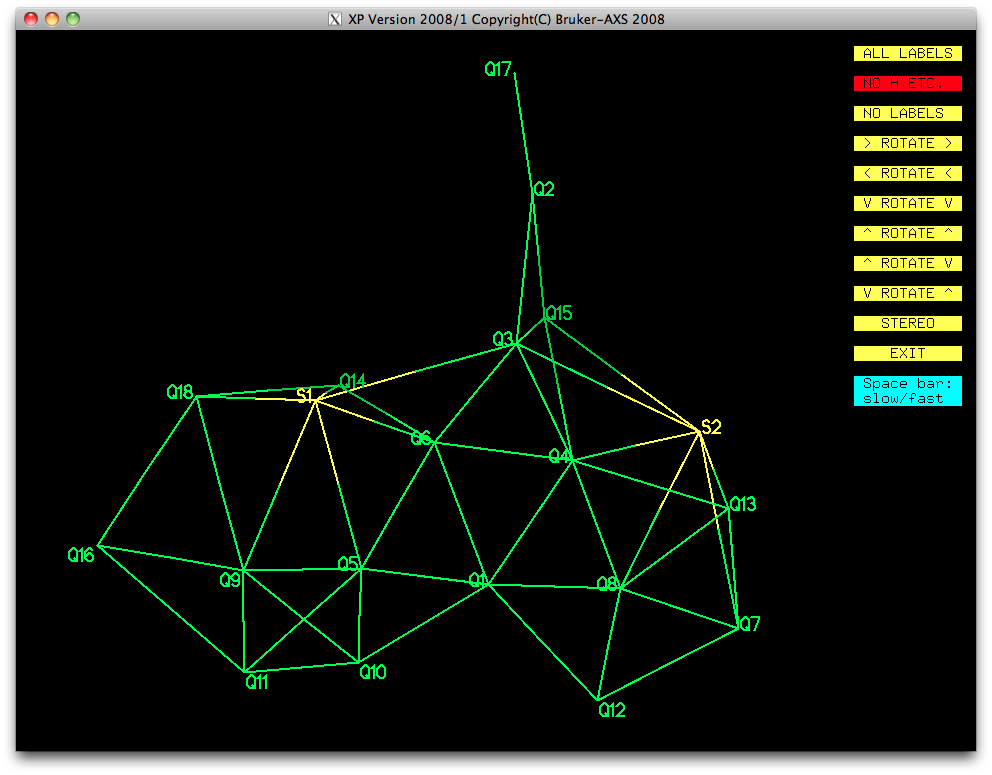
Since the likely structure was already known, combined with the fact that the higher
Q-numbered peaks are most likely to be spurious, some judicious picking gave the
following:
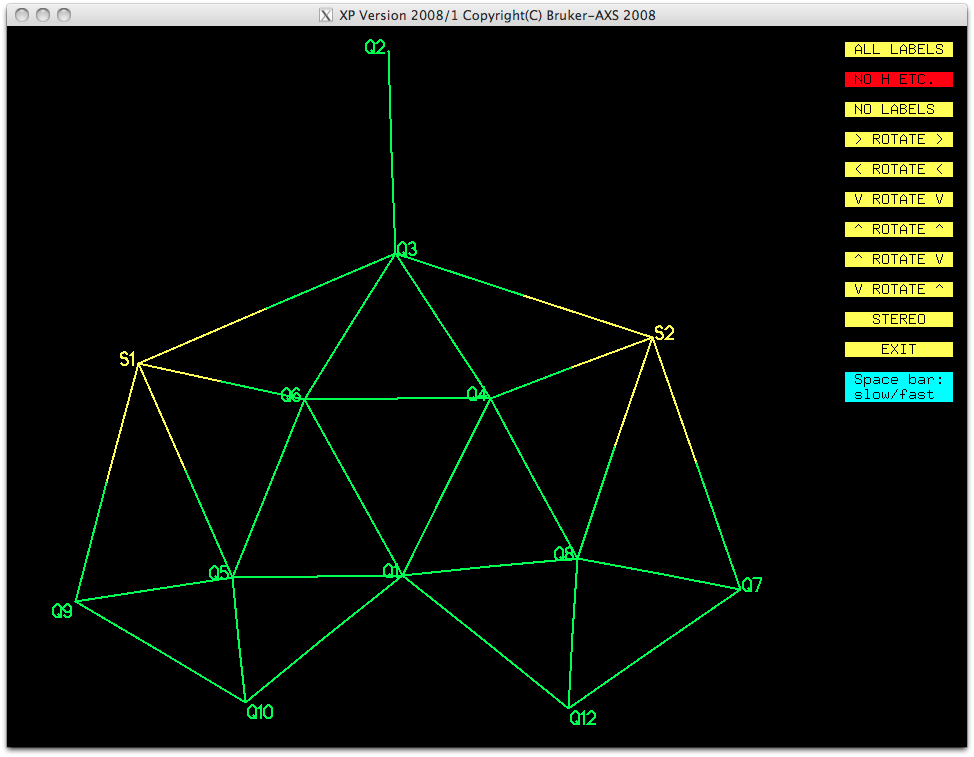
Although this does not look exactly like the suggested structure, it is possible
with some imagination and chemical intuition, to identify the gross structure of the
molecule in two orientations. Here's one orientation after numbering the atoms, but
leaving the other Q-peaks intact:
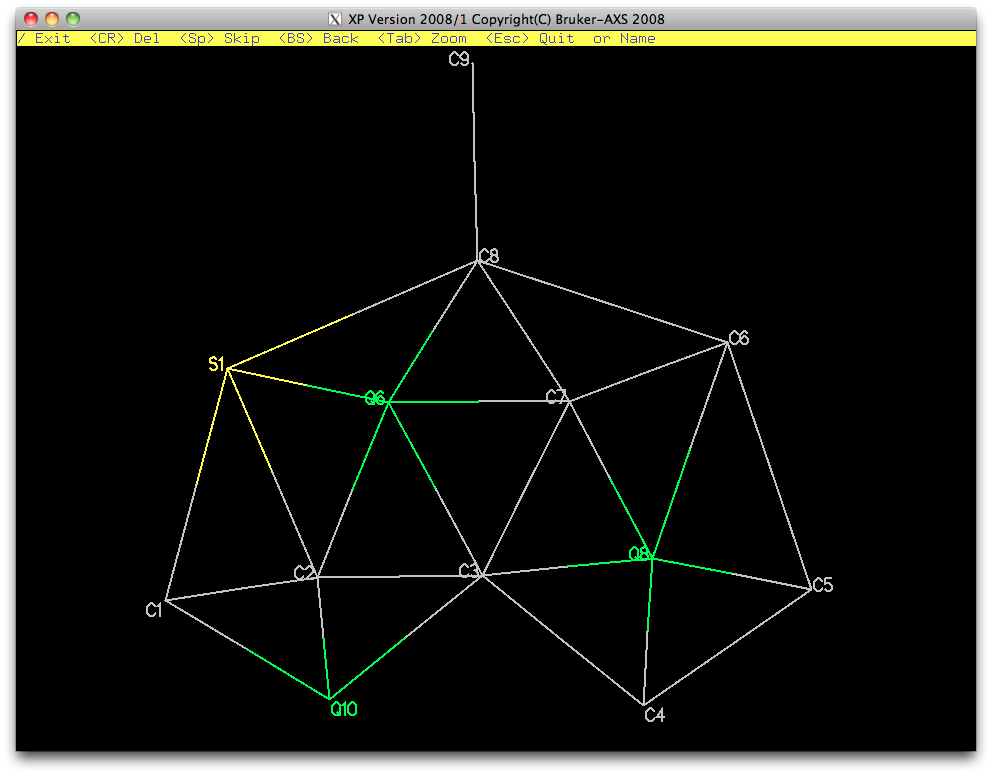
A view without the remaining Q-peaks (fmol less $q) looks like this:
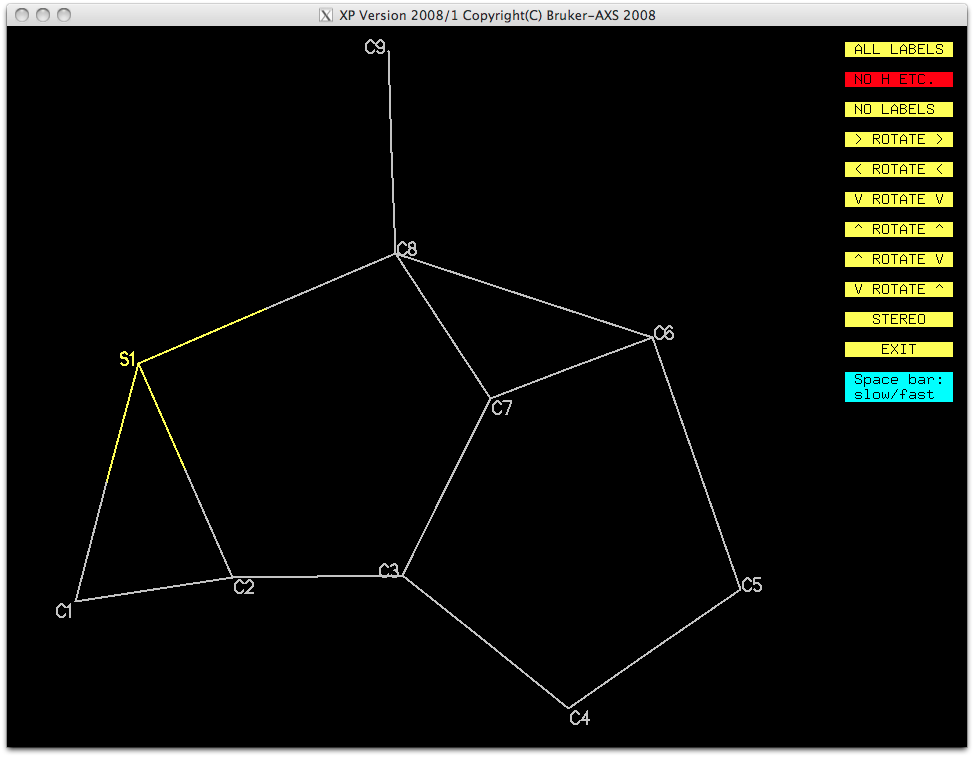
It's very distorted, but should be refinable, at least with some restraints. This
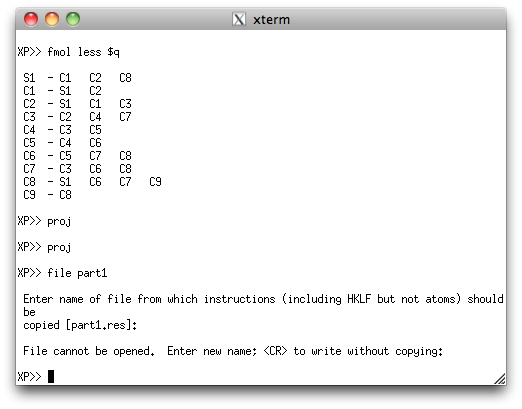
partial atom list can be sorted and filed for later use, as shown below.
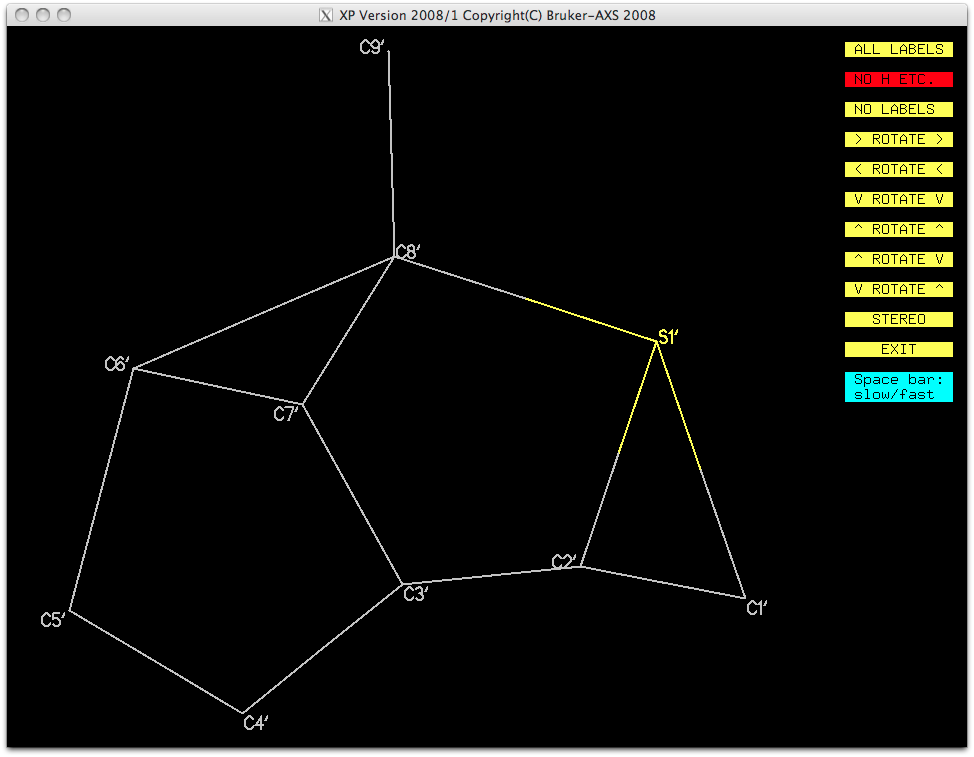
The other fragment can then be isolated and named in a similar way. It is important to
ensure that the numbering of the pieces follows some logical and related order. This
second orientation is no better looking than the first, but again, the gross structure
is visible.
This second PART is sorted and filed, just like the first PART.
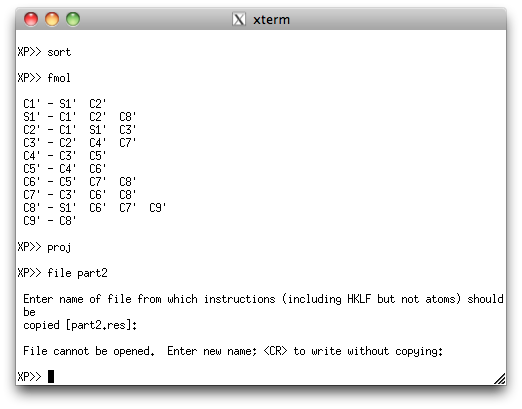
Note that XP gripes about not being able to open a file from which to get
instructions. That's not a problem, as all we want is the atom lists to cut/paste
into a new '.ins' file that we'll make by hand, next.
There are now a couple of incomplete '.ins' files named part1.ins and part2.ins that look like this ...
There are now a couple of incomplete '.ins' files named part1.ins and part2.ins that look like this ...
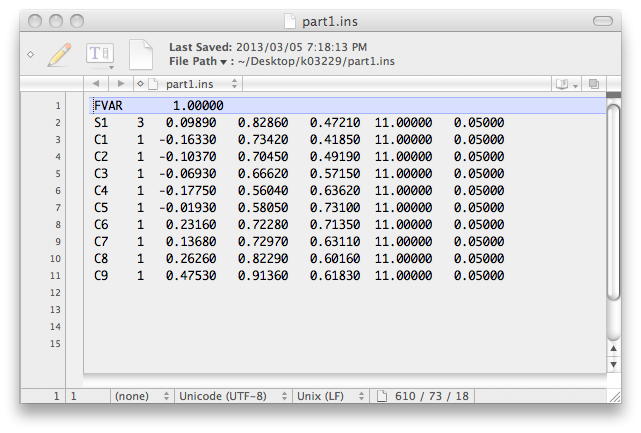
... and this:
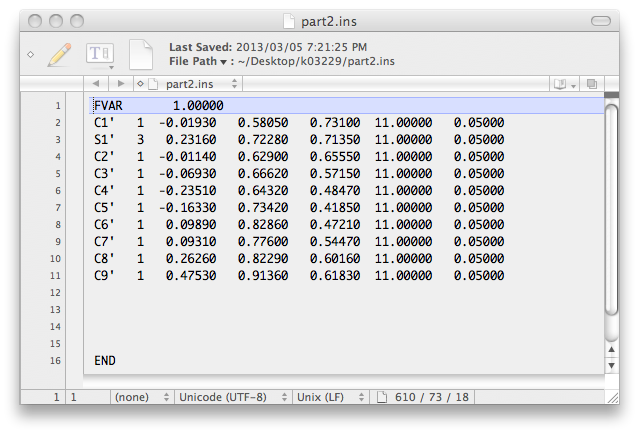
To incorporate these atom lists into the next '.ins' file, open up the previous '.res'
file with a text editor and replace the old atom list there with these two atom lists.
After some additional editing, this file should be saved as the next '.ins' file. It
ought to look something like this:
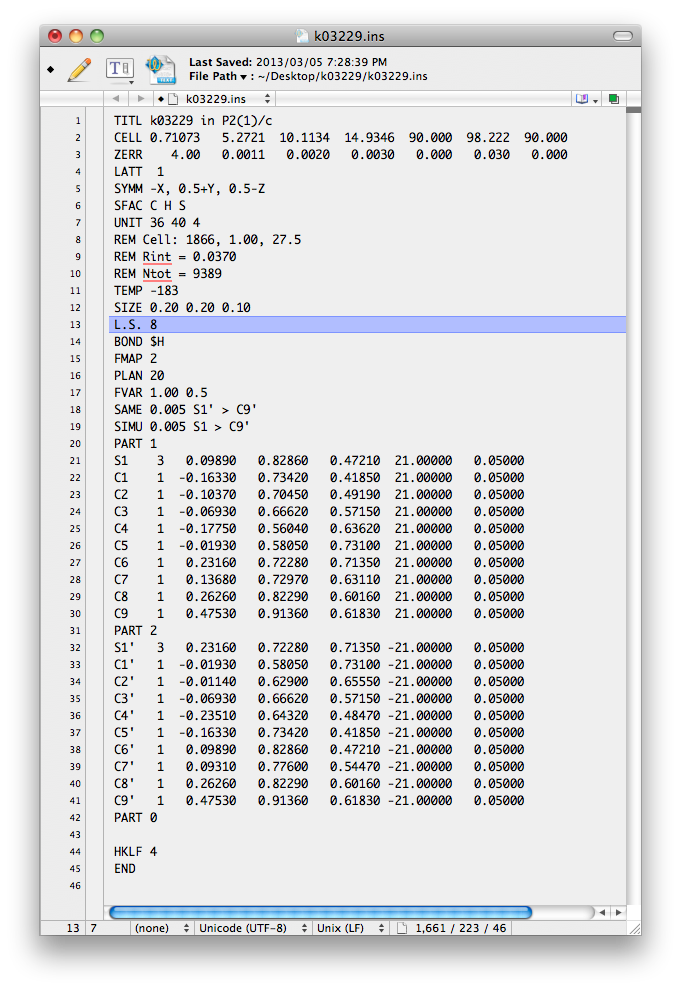
Here is a list of the changes made to the file.
1) Some comments (REM) added with information from nreport.html.
2) SAME 0.005 S1' > C9'
3) SIMU 0.005 S1 > C9'
4) FVAR 0.5 added.
5) Occupancies of PARTs set to 21.00000 and -21.00000
2) SAME 0.005 S1' > C9'
3) SIMU 0.005 S1 > C9'
4) FVAR 0.5 added.
5) Occupancies of PARTs set to 21.00000 and -21.00000
The order of the atoms is important. The SAME instruction tells SHELXL
that the geometry (1,2 and 1,3 distances) for the atom sequence S1' ... C9' is the same
as that for S1 ... C9. The SIMU instruction ensures that the Uiso
values for all the atoms are similar. At this stage, with such crummy geometry, these
restraints need to be quite strong (that's the 0.005 number), but will be loosened later.
Each part is given a fractional occupancy defined by free-variable number 2
(i.e. the second FVAR number), which has been set to 0.5 (a reasonable guess).
Note: If you are lucky enough to have a better looking, less distorted fragment, it can sometimes be simpler to use the SHELXL construct FRAG...FEND to place the second disorder fragment. An example of how to do this is given in a different tutorial.
In the next section, this model will be refined.
Note: If you are lucky enough to have a better looking, less distorted fragment, it can sometimes be simpler to use the SHELXL construct FRAG...FEND to place the second disorder fragment. An example of how to do this is given in a different tutorial.
In the next section, this model will be refined.
Return to the first page of this tutorial.