
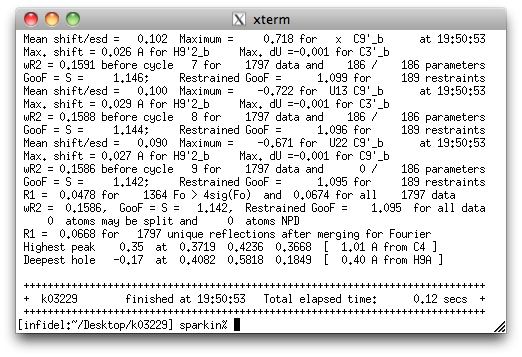
Notice that the wR2 value starts very high (~75%), but rapidly
plummets down to ~33% at the end. From the looks of it, it is probably not
completely converged, but at this stage that is not so important. It is also
worth noting that the difference map features i.e. the "Highest peak"
and "Deepest hole" indicate that there are no dramatically wrong or missing
atoms. To get a proper handle on whether the refinement run was a success,
a graphical view is a must. Here's what XP shows:
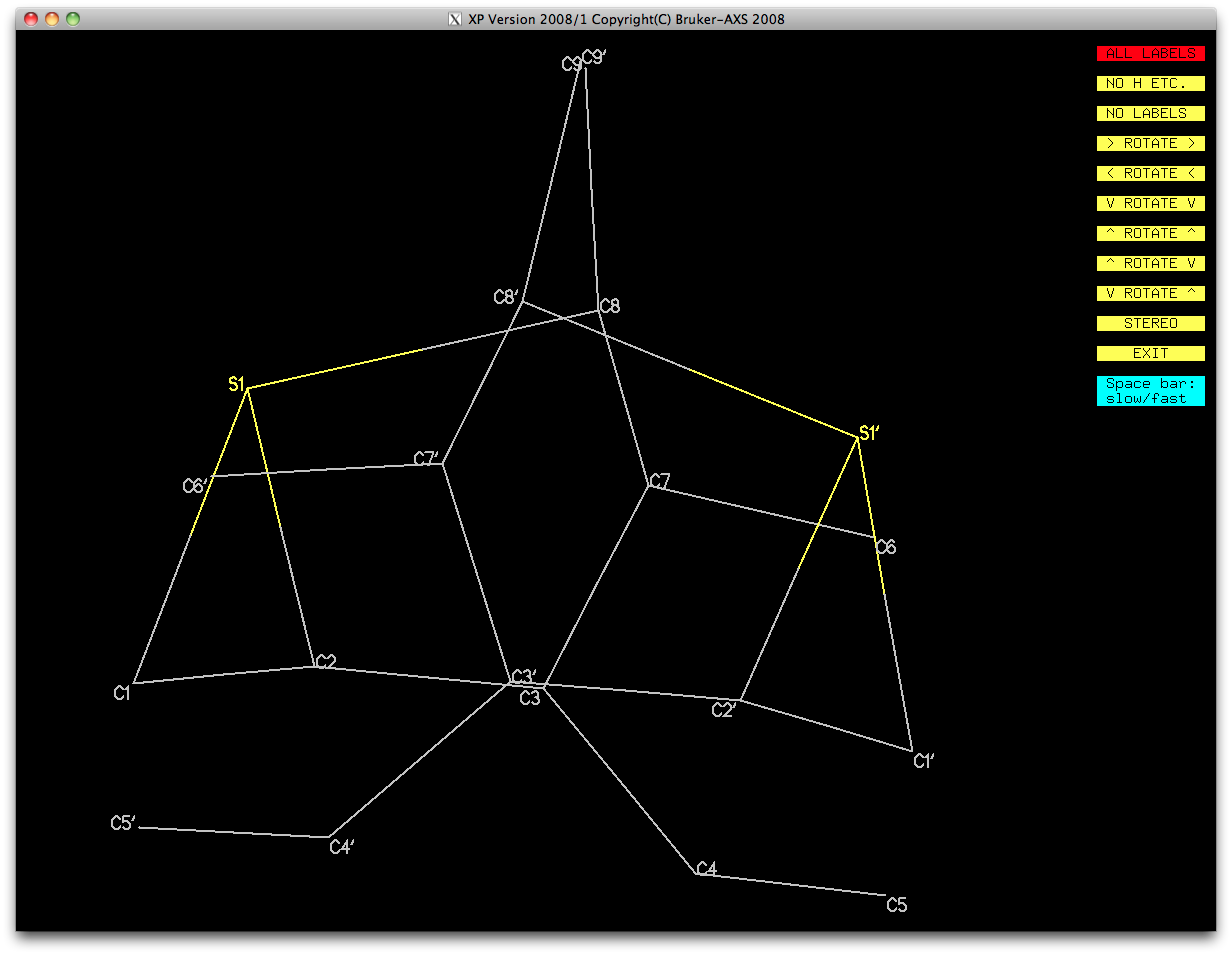
At first glance that looks pretty bad, but on closer inspection it is probably
fixable. In the last SHELXL run, the restraints tore the models apart
at the bottom, and created a pair of very distorted sulphur-containing rings.
It is well to remember that these programs do not know any chemistry. They
simply adjust parameters (x, y, z, and Uij,
etc.) in a least-squares fit to minimize the difference between
Fc2 and Fo2. In this
case, manual intervention is necessary to make it chemically sensible. The weird
S-containing rings, for example, can be fixed by swapping the coordinates of
C8 and C8'. In a similar way, switching C1 with C5' and C1' with C5 should
restore the connectivity of each PART, and highlight the improved
geometry over the starting model.
The editing is easily done by hand using cut/paste in a text editor, giving something like this:
The editing is easily done by hand using cut/paste in a text editor, giving something like this:
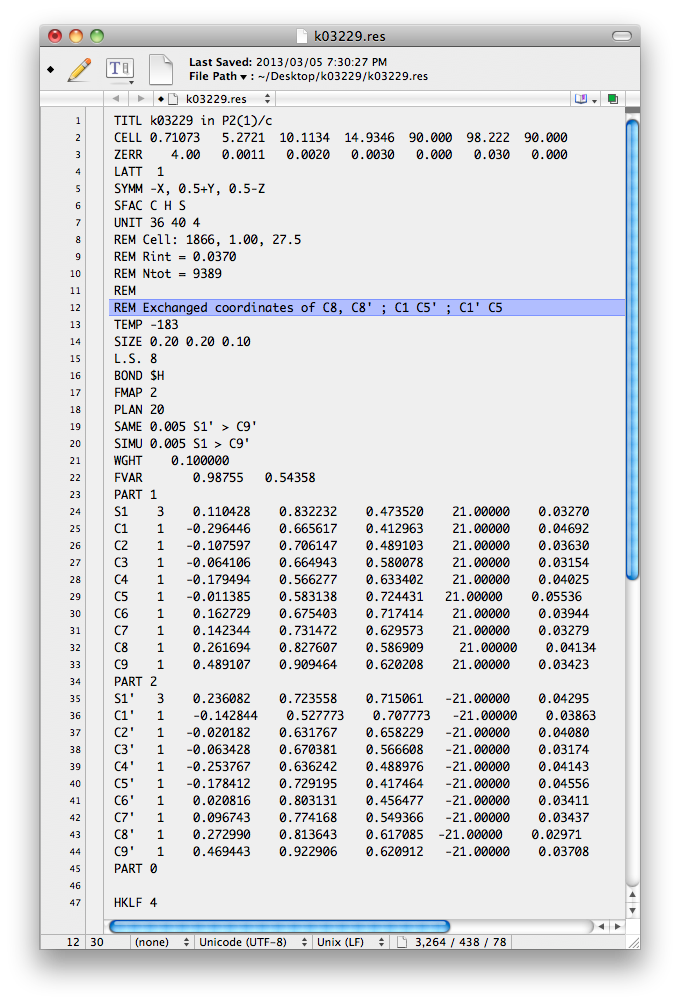
In the above image, the coordinates of the aforementioned atoms have been switched and
a REM comment added to explain what was done. The REM is not strictly
necessary, but it always helps to keep good notes, and the '.ins' file is a better place
than on scraps of easily lost paper. In any event, when filed as a new '.ins' file and
run through SHELXL again, the following is output to the screen.

The resulting geometry now looks pretty good (look at it with XP if you like).
It's time to think about adding hydrogen atoms and going anisotropic etc.
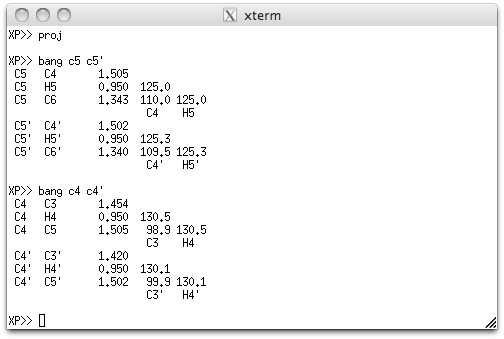
Before adding hydrogens, however, we need to know where the double bond is (or if it
is scrambled by additional disorder, as per the initial suspicion). In the XP
window above, the BANG command shows that C5=C6 (and C5'=C6') are double bonds,
while C4-C5 (and C4'-C5') are single. Often it is best to err on the side of caution,
so we'll run a refinement without adding the methylene-type hydrogens to C4 and C4'.
Thus, for the next refinement run the following HFIX commands go in the '.ins'
file:
HFIX 43 C5 C6 C5' C6'
HFIX 137 C1 C9 C1' C9'
HFIX 137 C1 C9 C1' C9'
It is possible with the disorder, that HFIX 137 will lead to non-convergence,
but if that is the case the '137' will be changed to '33' later. This refinement gives
the following ...
... which looks like this after removal of all but the four largest difference map peaks:
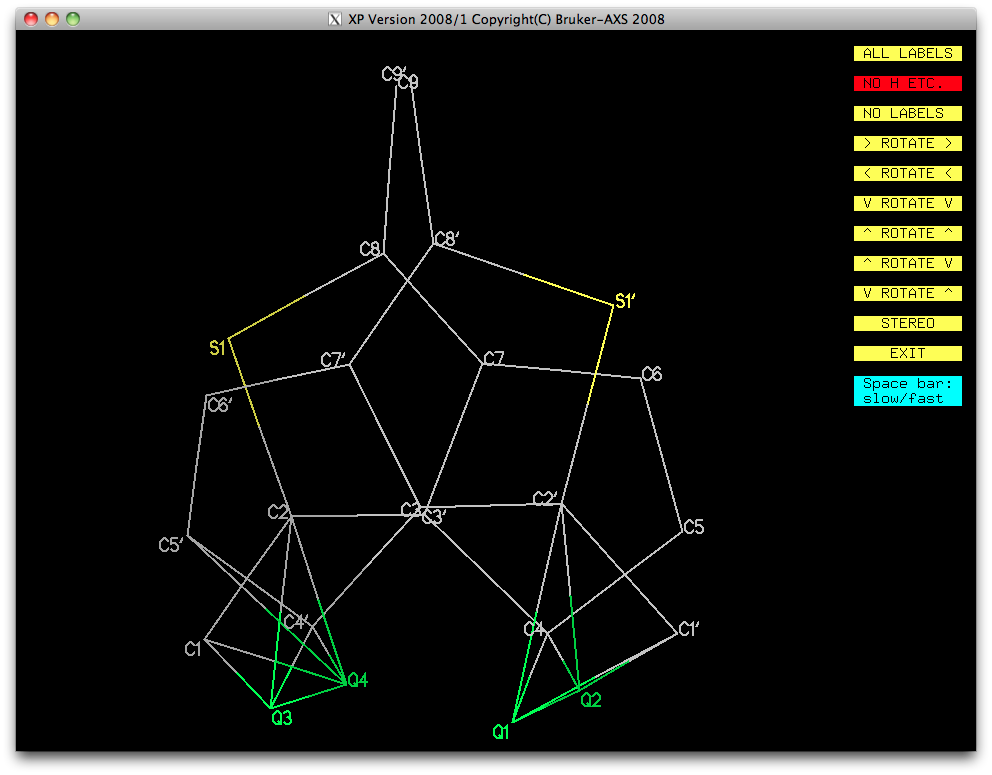
Those green Q-peaks look a lot like methylene hydrogen atom positions ...
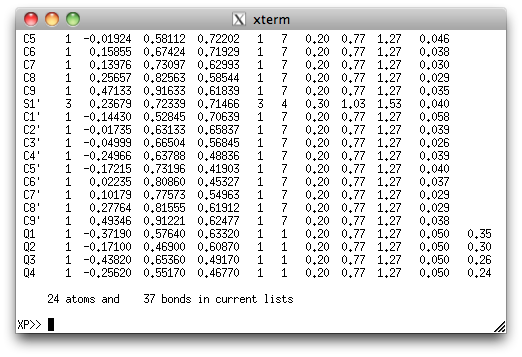
... in spite of their small size (0.24-0.35 eÅ-3). Subsequent
refinement with anisotropic displacement parameters (ANIS in the '.ins') gives
the following:
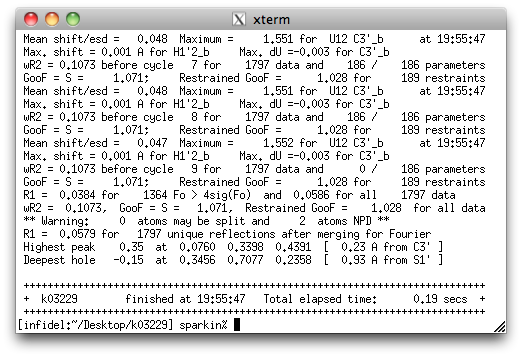
That's not all good news. SHELXL gives a warning about two atoms being 'NPD',
or non-positive definite. In plain language that means that the 'ellipsoids' for
those two atoms are not closed (i.e., they are not actually ellipsoids).
To proceed, we need to know which atoms are causing trouble. This is given in the '.lst'
file, which includes all sorts of useful diagnostics from the SHELXL run. Here's
the relevant part of the '.lst' file:
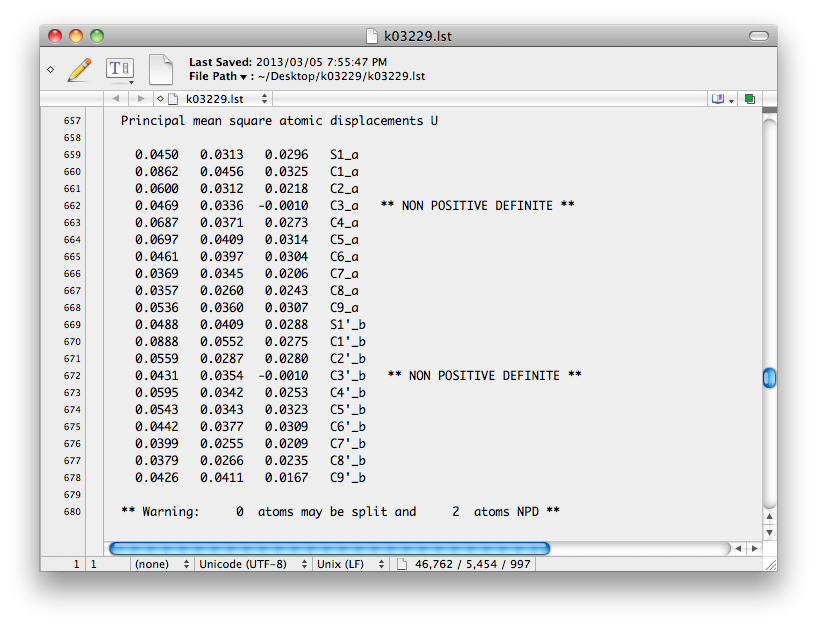
A quick look at the superimposed PARTs shows C3 and C3' virtually on top of each
other, so it is not in the least surprising that SHELXL couldn't refine separate
ADPs for these atoms. C9 and C9' are in similar danger. Atoms that overlap like this
need to have their ADPs fixed with EADP, or perhaps with very strong restraints.
This is also a good opportunity to relax and change the restraints on geometry and other
ADPs as well.
For the last rounds of refinement, the following changes were made:
For the last rounds of refinement, the following changes were made:
EADP C3 C3'
EADP C9 C9'
SAME 0.02 S1' > C9'
RIGU 0.01 S1 > C9'
ISOR 0.005 C3 C3'
EADP C9 C9'
SAME 0.02 S1' > C9'
RIGU 0.01 S1 > C9'
ISOR 0.005 C3 C3'
Here, RIGU is a new 'enhanced rigid bond' restraint in the latest release of
SHELXL. This refinement gave the following screen output.
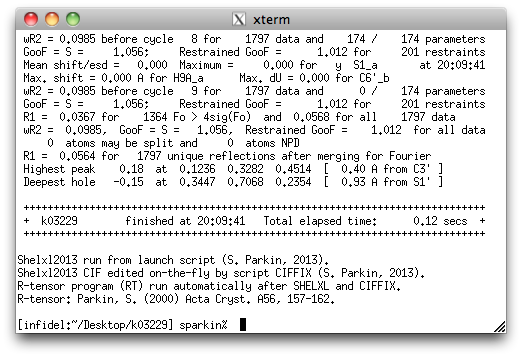
In the above, it is clear that the refinement has converged because the "Max. shift/esd"
is zero. The difference map is also quite flat, and other refinement statistics are
acceptable. The only things left are small tasks such as checking the weighting scheme
(WGHT), testing for possible extinction (EXTI), and ensuring that the
centroid of the molecule(s) are within the unit cell box. The latter is just for the
sake of aesthetics.
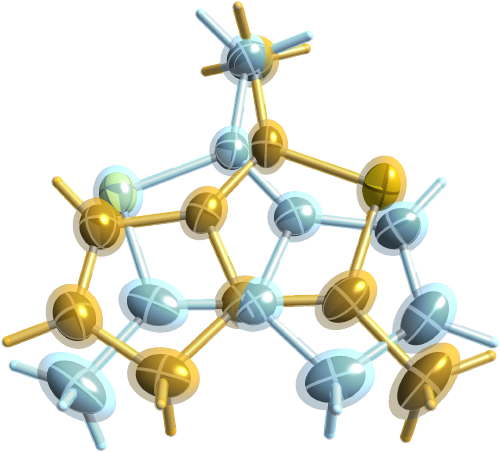
The final model, as drawn with shelXle is
shown above. It could be argued that the ADPs of the related atoms in the two
disordered parts should be constrained about the 2-fold rotation axis that relates them.
That would be nice, but such minutiae would make little difference to the model.
Return to the first page of this tutorial.