3) Other menial clean-up and book-keeping tasks.
Your data have now been extracted from the raw images, scaled and equivalent measurements have been merged. At this stage we can get a convenient summary of the data collection and processing as follows.
From a command line in a terminal shell, type "nreport" followed by the return key - like this:
You won't see much, just some message about retrieving cell information from the scalepack log files, viz.
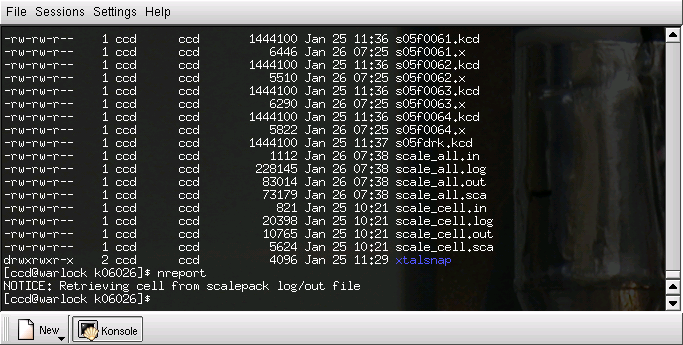
What "nreport" does is to write a summary file named "nreport.html" in (you guessed it) html format that can be read by a web-browser. It is probably wise to rename this file so that it corresponds to the sequence number of your data collection (in the terminal shell window type "mv nreport.html k06026.html" or similar).
To see what the nreport summary looks like, click here (it will open a new window)
The nreport summary contains a bunch of useful information. In particular, the number of reflections used to refine the cell dimensions and the theta range for these reflections. There is other useful stuff in this file too, including the number of reflections (overall, full and partial), data completeness (should be very close to 100%), average intensity, overall signal-to-noise ratio and overall R-merge values. It is useful to know these numbers because you will need to manually edit some of them into the CIF file at the very end of your structure determination.
The scalepack program writes a final processed data file named "scale_all.sca". While some of the structure solution programs can understand the format of this file, the most widely used structure refinement program cannot. Luckily there is a simple utility named "cifin" that will convert this file to the standard format used by the SHELX series of programs. For those that understand FORTRAN, this format is (3I4,2F8.2 or possibly 3I4,2F8.2,I4). To perform this conversion, type "cifin" followed by the return key in the terminal shell window:
A window like this will appear ...
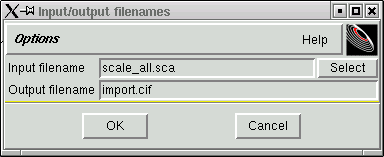
... change the "Output filename" to conform to your particular sequence number with the ".hkl" extension, viz.
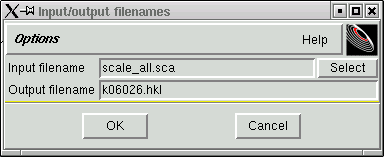
Click "OK" and a new file will be created that is in the format required by the SHELX programs.
Go on to absorption correction with SADABS (Note: this section is out of date).
Go back to the main Kappa data processing menu
Return to the main Tutorials page or to the main X-Ray Lab page