1) Get ready to solve the structure.
The last thing you did was to process the data, i.e. integrate, scale and merge all the intensities. This means that you should have a supergui window that looks something like this:
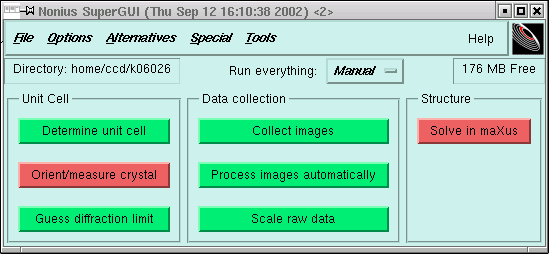
You need to click on the "Solve in maXus" button. When you do this you should get a small window like this:
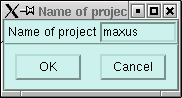
You can change the "name of the project" if you like, but there is usually no particular need to change it. When you click on the "OK" button here you will get the following:
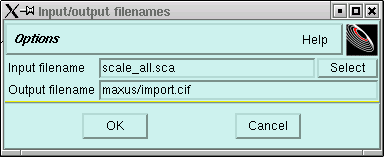
In the last step of the data processing stage, supergui used a program called "Scalepack", and this program wrote you a file with all the data in it called "scale_all.sca". Unfortunately the maXus programs cannot directly read this file. The window above comes to the rescue - it is a quick format conversion routine. It will reformat the data so that maXus can use them. In this case it will write you a file called "import.cif" with all the reflection data in "cif" format ("cif" stands for Crystallographic Information File). The conversion program is called "cifin", and it is possible to use it outside of maXus also. For instance, at some point you should use "cifin" to write a file for use by the SHELX programs (more on that later).
Anyway, we digress - do the obvious thing, click "OK" and you get this:
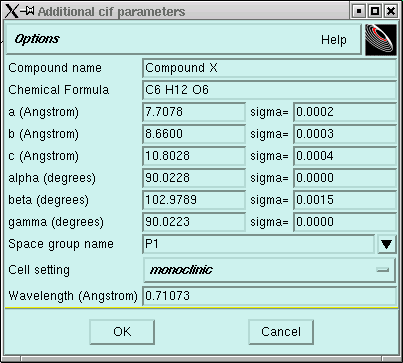
In this window you should change the "Compound name" so that it corresponds to your particular datafiles (i.e. k06123 etc.) If you can, it is a good idea to suggest a likely chemical formula. It does not matter if you get this formula exactly correct, but it does help to have the correct atom types in approximately the right proportion. Usually there will be little point in changing anything else. It does not matter at this stage if the space group is wrong because at this stage you haven't even tried to assign a space group. Nevertheless, if you do know something about the crystal, you might want to edit those items too, perhaps like this:
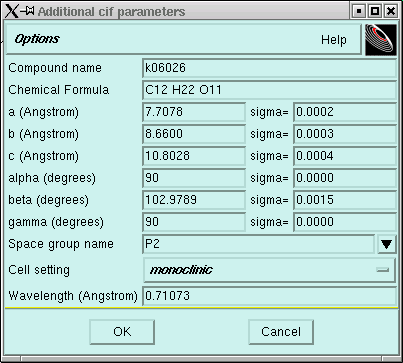
Notice here that the space group name was changed to P2. This is just the simplest monoclinic space group - it assumes nothing other than that the crystal system is monoclinic and the lattice is primitive. Notice also that because the crystal is monoclinic, alpha and gamma are both exactly 90° and there is no uncertainty attached to these 90° angles. In a monoclinic crystal two of the axes (by convention alpha and gamma) are mathematically exactly 90° - that's integer 90° - with no little bits here or there.
At some point you will tire of looking at this window and you will want to click "OK". Follow your impulse ...

... and this appears. Click on "OK" to proceed. If you don't do this the program will proceed on its own after a short pause and present you with this menu:
This is the maXus main menu. As stated earlier it is possible to use this menu to guide you through the whole process of structure determination right to the point of making nice pictures and preparing files for database deposition. Generally speaking we don't do that in this lab. but you are welcome to try, just don't expect much help beyond the menu item "Refine".
The sequence of steps that you use in maXus to get a viable structure solution are given in subsequent chapters of this tutorial.
Go on to maXus structure solution guide section 2
Go back to maXus structure solution guide main menu
Return to the main Tutorials page or to the main X-Ray Lab page